羟甲基甘氨酸钠溶液。
11.优选地,步骤(1)中的所述第一配制温度为44-46℃;所述步骤(2)中的所述第二配制温度为0-15℃。
12.优选地,步骤(3)中的所述第一进料温度为25-50℃;所述第二进料温度为0-15℃。
13.优选地,在所述步骤(3)中,通过调节所述进料泵的压力来分别调节物料a和物料b的流速,从而控制在所述连续流微反应器内的反应停留时间为15-30分钟。
14.优选地,在步骤(3)中,物料b中的甲醛和/或多聚甲醛折算成甲醛后的摩尔量为其理论摩尔量的1.05-1.08倍。
15.优选地,步骤(2)中,所述预定浓度的naoh溶液中,naoh的质量浓度为5-15%。
16.优选地,所述步骤(1)为:在第一配制温度下将甘氨酸和甘氨酸钠与水混合,得到物料a,其中,甘氨酸和甘氨酸钠的质量比为1:1.5-1:2.5。
17.优选地,所述步骤(2)为:在第二配制温度下将多聚甲醛与预定浓度的naoh溶液混合,得到物料b。
18.优选地,所述步骤(3)中的反应温度为40-60℃。
19.优选地,甘氨酸和甘氨酸钠的质量比为1:2。
20.本发明与现有技术对比的有益效果包括:
21.本发明通过改进合成工艺,获得一种能够稳定保存的n-羟甲基甘氨酸钠的高纯度结晶粉末状(核磁纯度≥95wt%),将其溶解配制的水溶液(浓度为20-50wt%)在保质期内(例如通常是3年)不会产生絮状悬浮物或其它漂浮物,且在保存一段时间后,该溶液中的n-羟甲基甘氨酸钠结晶析出后,仍可得到高纯度结晶粉末状(核磁纯度≥95wt%)。利用本工艺制备的高纯度结晶粉末状经过长时间的常温保存后,再次溶于水配置成浓度20-50wt%的水溶液,也不会产生絮状悬浮物、漂浮物或其它悬浊物,而且,本发明的连续流微反应器合成的n-羟甲基甘氨酸钠,批间差异小,且析晶制备成固体产品后,经过多次复检确认,具有很好的质量稳定性和均一性,预期其在两到三年的保质期内,仍可以很好的满足产品质量稳定性与均一性要求,因此,本发明制得的n-羟甲基甘氨酸钠解决了现有的产品合格率偏低的问题,能满足体外诊断行业检测试剂配制所需原料的质量要求。
附图说明
22.图1是本发明具体实施方式中的合成工艺流程图。
具体实施方式
23.下面对照附图并结合优选的实施方式对本发明作进一步说明。需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。
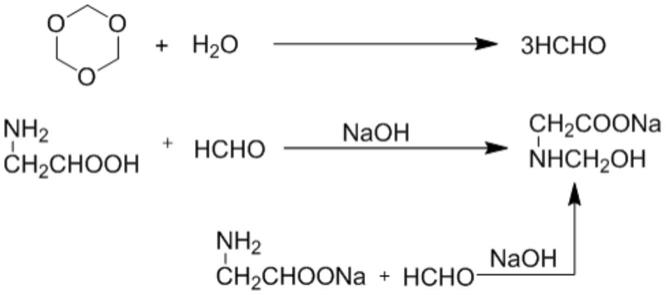
24.n-羟甲基甘氨酸钠的合成原理是将甘氨酸和/或甘氨酸钠与甲醛和/或多聚甲醛在氢氧化钠的存在下在水溶液体系中缩合而成,其反应方程式如下(其中,多聚甲醛以三聚甲醛为例,但并不限于三聚甲醛,其通常是聚合度大于2的各种多聚甲醛的混合物):
[0025][0026]
由于甲醛易挥发,原料无论是采用甲醛还是多聚甲醛,都会因为甲醛的挥发损耗而导致原料损失,如果完全按照摩尔比进行投料,就有可能因为甲醛的挥发而导致参与反应的甲醛的量低于理论值,从而导致甘氨酸和/或甘氨酸钠等原料不能反应完全,因此,为了提高甘氨酸和/或甘氨酸钠的转化率,现有技术采用的方案是,将甲醛和/或多聚甲醛的投料量过量,多余的甲醛可以通过后处理的溶剂脱除,而不影响产品的质量。但是,发明人经过多次实验后却发现,甲醛和/或多聚甲醛过量投料虽然可以提高转化率,但如果过量太多的话,则会产生副反应,导致产品中出现针状漂浮物或絮状悬浮物。因此,本发明的构思在于,将传统的釜式反应更改为连续流微反应器反应,可以降低甲醛的挥发损耗,并进一步优化物料的投料比,提高各原料转化率,提高目标产品的收率,并降低肉眼可见杂质的生成。
[0027]
在下文中,如无特别说明,“纯度”均指的“核磁纯度”。
[0028]
如图1所示,在一个具体实施方式中,n-羟甲基甘氨酸钠的连续流合成工艺包括如下步骤:
[0029]
(1)在第一配制温度下将甘氨酸和/或甘氨酸钠与水混合,得到物料a;
[0030]
(2)在第二配制温度下将甲醛和/或多聚甲醛与预定浓度的naoh溶液混合,得到物料b;
[0031]
(3)将第一进料温度下的物料a和第二进料温度下的物料b分别通过进料泵注入混合器内,然后进入连续流微反应器,在所述连续流微反应器内反应停留预定时间后,收集反应液;其中,在反应过程中,物料b中的甲醛和/或多聚甲醛折算成甲醛后的摩尔量为其理论摩尔量的1.01-1.1倍,且甲醛和/或多聚甲醛折算成甲醛后的摩尔量与naoh的摩尔量之比为1:1;
[0032]
(4)将所收集的反应液经过后处理后,得到固体产物,或得到20wt%-50wt%的n-羟甲基甘氨酸钠溶液。
[0033]
本发明采用连续流微反应的优势在于:1、各原料的投料量可精准控制,易于实现最佳物料配比;2、设备换热效果优异,能实现反应温度的精准控制;3、可以通过调节泵压和阀门开度控制进料液体流速,从而控制在反应器内的停留时间,精确控制反应时长;4、几乎没有混料返流现象,可减少副反应的发生。因此,本发明可以得到质量均一稳定、纯度较高的n-羟甲基甘氨酸钠的高纯度结晶粉末状。
[0034]
在优选的实施方式中,步骤(1)中的所述第一配制温度为44-46℃;所述步骤(2)中的所述第二配制温度为0-15℃。在44-46℃的配置温度下配制物料a可以使得甘氨酸和/或甘氨酸钠能更充分地溶解于水,从而减少溶剂水的用量,在0-15℃配制物料b可以尽可能地
避免naoh与hcho自身发生歧化反应而带入杂质。
[0035]
在优选的实施方式中,步骤(3)中的所述第一进料温度为25-50℃;所述第二进料温度为0-15℃。
[0036]
在优选的实施方式中,在所述步骤(3)中,通过调节所述进料泵的压力来分别调节物料a和物料b的流速,从而控制在所述连续流微反应器内的反应停留时间为15-30min,更优的是停留时间为22min。
[0037]
在优选的实施方式中,在步骤(3)中,物料b中的甲醛和/或多聚甲醛折算成甲醛后的摩尔量为其理论摩尔量的1.05-1.08倍。发明人经过多次对比实验发现,一个比较好的投料配比是,甲醛和/或多聚甲醛的投入量是其理论摩尔量的1.01-1.1之间,更优选的投料量是理论摩尔量的1.05-1.08,且如果以甘氨酸和/或甘氨酸钠设为一个单位摩尔量的投料值的话,氢氧化钠与甲醛和/或多聚甲醛的投料配比,维持在1:1的合成反应效果优于氢氧化钠按照理论摩尔量投料,例如,当物料a中含甘氨酸和甘氨酸钠两者,物料b中含多聚甲醛时,以甘氨酸和甘氨酸钠两者的摩尔量之和作为一个单位摩尔量,则多聚甲醛折算成甲醛后的摩尔量再加上若物料b中还含甲醛时甲醛的摩尔量之和与氢氧化钠的摩尔量之比为1:1。换句话说:只有甘氨酸和/或甘氨酸钠不过量,但甲醛和/或多聚甲醛与氢氧化钠均适量过量的投料配比,其合成效果优于三者按反应摩尔比的理论值进行投料,又经过多次对比实验发现一种较为理想的投料配比(按反应摩尔比)区间是:(甘氨酸和/或甘氨酸钠):(甲醛和/或多聚甲醛):氢氧化钠=1:1.05:1.05~1:1.08:1.08。
[0038]
在优选的实施方式中,步骤(2)中,所述预定浓度的naoh溶液中,naoh的质量浓度为5-15%。采用该浓度的naoh水溶液配制物料b,可以尽可能地避免naoh与hcho自身发生歧化反应而带入杂质。
[0039]
在优选的实施方式中,所述步骤(1)为:在第一配制温度下将甘氨酸和甘氨酸钠与水混合,得到物料a,其中,甘氨酸和甘氨酸钠的质量比为1:1.5-1:2.5。更有选的是甘氨酸和甘氨酸钠的质量比为1:2.
[0040]
在优选的实施方式中,所述步骤(2)为:在第二配制温度下将多聚甲醛与预定浓度的naoh溶液混合,得到物料b。
[0041]
在优选的实施方式中,所述步骤(3)中的反应温度为40-60℃。
[0042]
以下,通过实施例,对本发明做进一步详细说明。
[0043]
实施例1
[0044]
n-羟甲基甘氨酸钠的连续流合成工艺包括如下步骤:
[0045]
(1)将33.84gnaoh溶于160ml纯净水中,超声溶解,制得质量分数为17.5%的氢氧化钠溶液。
[0046]
(2)取120ml步骤(1)的氢氧化钠溶液加入反应瓶中,然后加入60.056g甘氨酸在45℃下搅拌,再加入120ml纯净水,得到约291ml甘氨酸钠/甘氨酸的混合水溶液,作为物料a。
[0047]
(3)取40ml步骤(1)的氢氧化钠溶液,加入80ml纯净水,得到120ml质量分数为7%的氢氧化钠溶液,在7%的氢氧化钠溶液中加入24.024g多聚甲醛,冰浴(0℃)下搅拌,再加入100ml纯净水超声溶解完全,得到约240ml naoh/hcho/多聚甲醛水溶液,作为物料b。
[0048]
(4)控制物料a的进料温度为25~50℃,物料b的进料温度为0~15℃,并设置连续流微反应器的反应温度为45℃,开启对物料a和物料b的连续进料,物料a和物料b分别通过
进料泵(如柱塞泵)分别进入混合器,经过混合器的快速混合后,进入连续流微反应器,经过内部换热后反应液的温度被精准的控制在45℃,且温度变化的上下幅度均不超过0.1℃,在经过预定的反应停留时间(15-30min)后,从连续流微反应器的出料口收集反应液。
[0049]
其中,通过调节两路进料泵(柱塞泵)的压力来调节流速,从而控制反应时长,本例中,物料a的进料流速为1.2125ml/min,物料b的进料流速为1.0ml/min,出料的反应液的ph为10.5。在实际生产中,该连续流微反应器还可以设有连续监控进料流速、进料温度、反应温度的功能,并可连续检测反应物料的ph值和红外谱图等监控指标的变化情况,从而更好的监控合成反应的原料转化率等变动情况。
[0050]
(5)将所收集的反应液经过后处理后,得到固体产物。其中,后处理过程为:将反应液旋蒸浓缩体积,到其成为白色油状液体,向反应液中加入80ml无水乙醇,不断搅拌除水,待溶液分层后,下层呈现白色油状液体,分液将上层清液倒掉,继续加入80ml无水乙醇,分液,在尚未明显析晶之前用滤纸过滤去除不溶物,重复此步骤2-3次,待有固体析出后,抽滤,将固体产品进行常温真空旋转干燥,得到固体产物约90克。得到的固体产物还可以进一步重结晶后进一步提纯。根据需要,得到的固体产物可以直接保存,也可以再次溶于水后得到20wt%-50wt%的n-羟甲基甘氨酸钠溶液进行保存。
[0051]
在本例中,得到的固体产物的质量收率为96%,核磁纯度为96%,收率92.2%(收率=质量收率*核磁纯度),将该固体产物经过一次重结晶后可提纯到99%以上(本例中,经过一次重结晶后得到的产物的核磁纯度为99.4%),得到提纯后的固体产物。用提纯后的固体产物配制质量分数为30%的水溶液无明显悬浮物,符合工艺要求。提纯后的固体产物的相关检测报告如下表1所示:
[0052]
表1
[0053]
[0054][0055]
注:表1中的杂质元素通常来自上游原料,如氢氧化钠、甘氨酸、甘氨酸钠、甲醛、多聚甲醛、水等,且通过紫外光谱a280nm/a260nm实验表明最终产物中几乎不含蛋白、酚类等杂质。
[0056]
比较例1
[0057]
n-羟甲基甘氨酸钠的釜式反应合成工艺,包括如下步骤:
[0058]
(1)将8.46gnaoh溶于40ml去离子水中,超声溶解,配制为质量分数为17.5%的氢氧化钠溶液。
[0059]
(2)取30ml步骤(1)的氢氧化钠溶液加入反应瓶中,然后加入15.014g甘氨酸在45℃下搅拌,得到物料a。
[0060]
(3)取10ml步骤(1)的氢氧化钠溶液,加入10ml纯净水,得到20ml质量分数为10%的氢氧化钠溶液,在10%的氢氧化钠溶液中加入6.006g多聚甲醛,冰浴(0℃)下搅拌,超声溶解完全,得到物料b。
[0061]
(4)将物料a和物料b加入反应釜内,在45℃下搅拌反应6h。
[0062]
(5)将反应结束后收集的反应液进行后处理后,得到固体产物。其中,后处理过程为:将反应液旋蒸浓缩体积,到其成为白色油状液体,向反应液中加入50ml无水乙醇,不断搅拌至少10分钟,再静置10分钟待溶液分层后,下层呈现白色油状液体,分液将上层清液倒掉,继续加入70ml无水乙醇,分液,重复此步骤2-3次,待有固体析出后,抽滤,将固体产品干燥得到23g固体产物,其质量收率为95.37%,核磁纯度为94%,收率为89.65%,将该固体产物经过一次重结晶后可提纯到98%以上(本例中,经过一次重结晶后得到的产物的核磁纯度为98.5%),得到提纯后的固体产物。
[0063]
分别按照实施例1的步骤和比较例1的步骤各重复10次实验,比较不同批次的产物的检测结果,如下表2和表3所示,其中,核磁纯度是指得到的固体产物经过一次重结晶后的产物的纯度,浊度采用浊度仪(型号:zxzd-2008)测得,澄清度的检测方法为:在暗处从底部往上打光,通过溶液(20-50wt%的水溶液)中是否有微粒散射或出现阴影等现象,经质检人员肉眼观察判断该水溶液是否有絮状物、漂浮物(通常,目测发现有絮状物或漂浮物,则判定为不合格)。
[0064]
表2:n-羟甲基甘氨酸钠(按实施例1的连续流合成工艺)不同批次的检测结果
[0065][0066]
表3:n-羟甲基甘氨酸钠(按比较例1的釜式反应合成工艺)不同批次的检测结果
[0067][0068]
从表2和表3的对比可知,本发明实施例的产品纯度高,多次检测中浊度和澄清度的结果表明产品均有很好的质量稳定性和均一性,而釜式反应的产品具有较低的质量稳定性和均一性,不满足要求。
[0069]
通过上述实施例和比较例可知,本发明的优点包括如下:
[0070]
1、与传统的釜式反应相比,本发明可以缩短反应时长,能更加精准地控制反应温度及各项工艺技术参数,提高了反应的收率和产品质量的一致性与稳定性,降低了批次间差异,极大的减少了产品水溶液中的悬浮物。能更加精准的合成n-羟基甘氨酸钠,且副反应更少,产品纯度更高。经过试验证明,本发明的连续流微反应工艺,其核磁纯度可达95%以上,经过一次重结晶后核磁纯度可达99%以上,收率可达90%以上,均高于传统釜式反应,能实现连续化大批量生产。
[0071]
2、本发明的工艺技术路线,因副反应减少,产品的杂质少,长时间也保存不会产生明显的絮状物悬浮物或针状漂浮物;
[0072]
3、本发明生产的产品,不同批次直接的产品批间差很小,质量稳定,产量易于放大,能更好的满足下游客户对产品质量的严苛要求;
[0073]
4、本发明得到的无色透明液体中无肉眼可见杂质,析晶后得到白色结晶粉末,产品纯度更高且产品颜色较好,将白色结晶产品再次溶于水,配制成20wt%-50wt%的水溶液,也无肉眼可见杂质。
[0074]
以上内容是结合具体的优选实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属技术领域的技术人员来说,在不脱离本发明构思的前提下,还可以做出若干等同替代或明显变型,而且性能或用途相同,都应
当视为属于本发明的保护范围。