1.本发明属于深蓝光材料技术领域。更具体地,涉及一种氮杂环咔唑类化合物及其应用。
背景技术:
2.有机发光二极管因其自身重量轻、柔软性好、反应时间短、可视角宽、工作温度范围宽、亮度和对比度高等优点,逐步成为学者们广泛关注研究的焦点。经过几十年的发展,有机发光二极管的相关技术日益成熟,广泛用于全彩显示和白光照明两个领域,尤其是在平板显示、固体发光等领域的显示技术产品更加广受好评。
3.有机发光材料是有机发光二极管中的核心部分,因此有机发光材料的发射亮度、荧光量子产率等性质都对其性能产生直接的影响。现代有机合成技术已实现通过分子的设计、修饰来调节材料的发光性能,而选择或设计发光材料必须满足以下4点要求:1)荧光量子产率较高,无明显的猝灭现象;2)成膜性能良好,在制成膜时不产生针孔;3)载流子传输性能良好,即具有较高的导电率,能较好地传输电子;4)具有良好的热稳定性。
4.目前,已有研究制备得到含有氮杂咔唑-咪唑单元的有机发光材料和基于吖啶-氮杂咔唑单元的有机发光材料,这些均体现出氮杂环咔唑类化合物具有制备成为发光材料的潜力,但现如今氮杂环咔唑类化合物种类尚少,因此,迫切需要开发出更多新型的适用于制备发光材料的氮杂环咔唑类化合物。
技术实现要素:
5.本发明旨在提供一种氮杂环咔唑类化合物,为发光材料的制备提供一种新型原料选择。
6.本发明的另一目的是提供上述氮杂环咔唑类化合物在制备发光材料或发光器件中的应用。
7.本发明的又一目的是提供一种发光材料。
8.本发明上述目的通过以下技术方案实现:
9.本发明提供了一种氮杂环咔唑类化合物,所述氮杂环咔唑类化合物具有如下式(a1)或式(a2)所示的化学结构:
[0010][0011]
优选地,所述式(a1)化合物的制备方法包括如下步骤:
[0012]
s1.3,6-二叔丁基-9h-咔唑通过乌尔曼反应,得到中间体m1;
[0013]
s2.步骤s1所得中间体m1与2,4-联苯-6-(4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)苯基)-1,3,5-三嗪通过铃木反应,得到所述式(a1)化合物。
[0014]
进一步优选地,所述乌尔曼反应为:3,6-二叔丁基-9h-咔唑、1-溴-4-碘苯、碘化亚铜、碱性试剂在有机溶剂、惰性氛围中加热反应。
[0015]
更优选地,所述3,6-二叔丁基-9h-咔唑、1-溴-4-碘苯、碘化亚铜、碱性试剂的摩尔比为1:1.2~1.5:0.2~0.3:2~3。最优选为1:1.2:0.2:2,见实施例1。
[0016]
更优选地,所述碱性试剂为碳酸钾或碳酸钠。
[0017]
更优选地,所述有机溶剂为n,n-二甲基甲酰胺、n,n-二甲基乙酰胺或二甲基亚砜。
[0018]
更优选地,所述惰性氛围包括氦气、氮气或氩气。
[0019]
更优选地,所述加热为在≤140℃下加热12~15h。最优选为在140℃下加热12h。
[0020]
更优选地,所述加热反应后还通过冷却、萃取、脱溶剂、柱层析进行后处理,具体为:冷却,用乙酸乙酯萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚)得到所述中间体m1(9-(4-溴苯基)-3,6-二叔丁基-9h-咔唑)。
[0021]
作为一种可实施方案,所述乌尔曼反应的反应式为:
[0022][0023]
进一步优选地,所述铃木反应为:中间体m1、2,4-联苯-6-(4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)苯基)-1,3,5-三嗪、四(三苯基膦)钯、碱性试剂在有机溶剂、惰性氛围中回流反应。
[0024]
更优选地,所述中间体m1、2,4-联苯-6-(4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)苯基)-1,3,5-三嗪、四(三苯基膦)钯、碱性试剂的摩尔比为1:1~1.2:0.04~0.06:2~3。最优选为1:1.05:0.047:2.51,见实施例1。
[0025]
更优选地,所述碱性试剂为碳酸钾或碳酸钠。
[0026]
更优选地,所述有机溶剂为四氢呋喃。
[0027]
更优选地,所述惰性氛围包括氦气、氮气或氩气。
[0028]
更优选地,所述回流为在70~85℃下回流10~14h。最优选为在80℃下回流12h。
[0029]
更优选地,所述回流反应后还通过冷却、过滤、萃取、脱溶剂、柱层析进行后处理,具体为:冷却,过滤,用二氯甲烷萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚/二氯甲烷=1:1(v/v))得到所述式(a1)化合物(3,6-二叔丁基-9-(4'-(4,6-二苯基-1,3,5-三嗪-2-基)-[1,1'-联苯]-4-基)-9h-咔唑)。
[0030]
作为一种可实施方案,所述铃木反应的反应式为:
[0031][0032]
优选地,所述式(a2)化合物的制备方法包括如下步骤:
[0033]
s1.3,6-二叔丁基-9h-咔唑通过乌尔曼反应,得到中间体m2;
[0034]
s2.步骤s1所得中间体m2通过闭环反应,得到中间体m3;
[0035]
s3.步骤s2所得中间体m3与2,4-联苯-6-(4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)苯基)-1,3,5-三嗪通过铃木反应,得到所述式(a2)化合物。
[0036]
进一步优选地,所述乌尔曼反应为:3,6-二叔丁基-9h-咔唑、5-溴-2-碘苯甲酸甲酯、碘化亚铜、碱性试剂在有机溶剂、惰性氛围中加热反应。
[0037]
更优选地,所述3,6-二叔丁基-9h-咔唑、5-溴-2-碘苯甲酸甲酯、碘化亚铜、碱性试剂的摩尔比为1:1.2~1.5:0.2~0.3:2~3。最优选为1:1.2:0.2:2,见实施例2。
[0038]
更优选地,所述碱性试剂为碳酸钾或碳酸钠。
[0039]
更优选地,所述有机溶剂为n,n-二甲基甲酰胺、n,n-二甲基乙酰胺或二甲基亚砜。
[0040]
更优选地,所述惰性氛围包括氦气、氮气或氩气。
[0041]
更优选地,所述加热为在≤140℃下加热12~15h。最优选为在140℃下加热12h。
[0042]
更优选地,所述加热反应后还通过冷却、萃取、脱溶剂、柱层析进行后处理,具体为:冷却,用乙酸乙酯萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚)得到所述中间体m2(5-溴-2-(3,6-二叔丁基-9h-咔唑-9-基)苯甲酸甲酯)。
[0043]
作为一种可实施方案,所述乌尔曼反应的反应式为:
[0044][0045]
进一步优选地,所述闭环反应为:将中间体m2、苯基溴化镁在有机溶剂、惰性氛围
中加热反应,萃取、脱溶剂后溶解于反应溶液中,再加入酸性试剂进行回流。
[0046]
更优选地,所述中间体m2、苯基溴化镁、酸性试剂的摩尔比为5~6:1:1.5~3。最优选为5.03:1:2,见实施例2。
[0047]
更优选地,所述有机溶剂为四氢呋喃、乙醚或甲苯。
[0048]
更优选地,所述惰性氛围包括氦气、氮气或氩气。
[0049]
更优选地,所述加热为在50~60℃下加热4~5h,最优选为50℃下加热4h。
[0050]
更优选地,所述反应溶液为乙酸。
[0051]
更优选地,所述酸性试剂为盐酸或氢碘酸。
[0052]
更优选地,所述回流为在110~130℃下回流3~5h。最优选为在120℃下回流4h。
[0053]
更优选地,所述萃取采用二氯甲烷和水进行萃取。
[0054]
更优选地,所述脱溶剂为收集有机层并用na2so4干燥。
[0055]
更优选地,所述回流后还通过冷却、萃取、脱溶剂、柱层析进行后处理,具体为:冷却,用二氯甲烷和水萃取,蒸发有机层后通过柱层析法(洗脱剂为石油醚)得到所述中间体m3(10-溴-3,6-二叔丁基-8,8-二苯基-8h-吲哚[3,2,1-去]吖啶)。
[0056]
作为一种可实施方案,所述闭环反应的反应式为:
[0057][0058]
进一步优选地,所述铃木反应为:将中间体m3、2,4-联苯-6-(4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)苯基)-1,3,5-三嗪、四(三苯基膦)钯、碱性试剂在有机溶剂、惰性氛围中回流反应。
[0059]
更优选地,所述中间体m3、2,4-联苯-6-(4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)苯基)-1,3,5-三嗪、四(三苯基膦)钯、碱性试剂的摩尔比为1:1~1.2:0.04~0.06:2~3。最优选为1:1.05:0.047:2.51,见实施例2。
[0060]
更优选地,所述碱性试剂为碳酸钾或碳酸钠。
[0061]
更优选地,所述有机溶剂为四氢呋喃。
[0062]
更优选地,所述惰性氛围包括氦气、氮气或氩气。
[0063]
更优选地,所述回流为在70~85℃下回流10~14h。最优选为在80℃下回流12h。
[0064]
更优选地,所述加热反应后还通过冷却、过滤、萃取、脱溶剂、柱层析进行后处理,具体为:冷却,过滤,用二氯甲烷萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚/二氯甲烷=1:1(v/v))得到所述式(a2)化合物(3,6-二叔丁基-10-(4-(4,6-二苯基-1,3,5-三嗪-2-基)苯基)-8,8-二苯基-8h-吲哚[3,2,1-de]吖啶)。
[0065]
作为一种可实施方案,所述铃木反应的反应式为:
[0066][0067]
作为一种优选地可实施方案,式(a1)化合物的制备包括如下步骤:
[0068]
s1.3,6-二叔丁基-9h-咔唑、1-溴-4-碘苯、碘化亚铜、碱性试剂(碳酸钾或碳酸钠)按1:1.2~1.5:0.2~0.3:2~3的摩尔比在有机溶剂(n,n-二甲基甲酰胺、n,n-二甲基乙酰胺或二甲基亚砜)、惰性氛围、≤140℃下加热12~15h后,冷却,用乙酸乙酯萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚)得到中间体m1;
[0069]
s2.步骤s1所得中间体m1、2,4-联苯-6-(4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)苯基)-1,3,5-三嗪、四(三苯基膦)钯、碱性试剂(碳酸钾或碳酸钠)按1:1~1.2:0.04~0.06:2~3的摩尔比在有机溶剂(四氢呋喃)、惰性氛围、70~85℃下回流10~14h,冷却,过滤,用二氯甲烷萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚/二氯甲烷=1:1(v/v))得到所述式(a1)化合物。
[0070]
作为一种优选地可实施方案,式(a2)化合物的制备包括如下步骤:
[0071]
s1.3,6-二叔丁基-9h-咔唑、5-溴-2-碘苯甲酸甲酯、碘化亚铜、碱性试剂(碳酸钾或碳酸钠)按1:1.2~1.5:0.2~0.3:2~3的摩尔比在有机溶剂(n,n-二甲基甲酰胺、n,n-二甲基乙酰胺或二甲基亚砜)、惰性氛围、≤140℃下加热12~15h后,冷却,用乙酸乙酯萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚)得到中间体m2;
[0072]
s2.将中间体m2、苯基溴化镁在有机溶剂(四氢呋喃、乙醚或甲苯)、惰性氛围、50~60℃下加热4~5h,加入二氯甲烷和水萃取、收集有机层并用na2so4干燥后溶解于反应溶液(乙酸)中,再加入酸性试剂(盐酸或氢碘酸),在110~130℃下回流3~5h后,冷却,加入二氯甲烷和水萃取,蒸发有机层后通过柱层析法(洗脱剂为石油醚)得到所述中间体m3;其中,所述中间体m2、苯基溴化镁、酸性试剂的摩尔比为5~6:1:1.5~3。
[0073]
s3.将中间体m3、2,4-联苯-6-(4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)苯基)-1,3,5-三嗪、四(三苯基膦)钯、碱性试剂(碳酸钾或碳酸钠)按1:1~1.2:0.04~0.06:2~3的摩尔比在有机溶剂(四氢呋喃)、惰性氛围、70~85℃下回流10~14h后,冷却,过滤,用二氯甲烷萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚/二氯甲烷=1:1(v/v))得到所述式(a2)化合物。
[0074]
上述氮杂环咔唑类化合物具有可作为深蓝光材料来源的卓越性能,如具有良好的热稳定性、较优的成膜性、较强的发射亮度、较高的荧光量子产率、较佳的传输电子能力,作为一种成本低、性能佳的新型有机发光小分子,十分适合制备成发光材料或发光器件,因此,上述氮杂环咔唑类化合物在制备发光材料或发光器件中的应用,以及包含上述氮杂环咔唑类化合物或由上述氮杂环咔唑类化合物制备而成的发光材料也应在本发明的保护范
围之内。
[0075]
本发明具有以下有益效果:
[0076]
本发明的氮杂环咔唑类化合物具有良好的热稳定性(热分解温度分别高达400℃和420℃)、较优的成膜性(玻璃化转变温度分别为120℃和150℃)、较强的发射亮度(分别产生188nm和145nm的红移)、较高的荧光量子产率(荧光量子产率分别为80.78%和65.14%)、较佳的传输电子能力(lumo分别为-2.48ev和-2.58ev),且该氮杂环咔唑类化合物制备原料来源广泛,价格低廉,合成条件温和,操作简单。
附图说明
[0077]
图1是实施例1所得产物a1的核磁共振氢谱图。
[0078]
图2是实施例2所得产物a2的核磁共振氢谱图。
[0079]
图3是实施例1所得产物a1的质谱图。
[0080]
图4是实施例2所得产物a2的质谱图。
[0081]
图5是实施例1所得产物a1的氧化电位图。
[0082]
图6是实施例2所得产物a2的氧化电位图。
[0083]
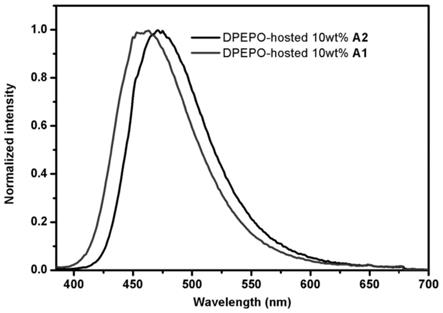
图7是实施例1~2所得产物的热重分析图(td)。
[0084]
图8是实施例1~2所得产物的玻璃化转变温度图(tg)。
[0085]
图9是实施例1所得产物a1的溶剂化效应图。
[0086]
图10是实施例2所得产物a2的溶剂化效应图。
[0087]
图11是实施例1所得产物a1荧光强度变化的荧光光谱图。
[0088]
图12是实施例2所得产物a2荧光强度变化的荧光光谱图。
[0089]
图13是实施例1~2所得产物的薄膜荧光发射图。
具体实施方式
[0090]
以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
[0091]
除非特别说明,以下实施例所用试剂和材料均为市购。
[0092]
实施例1一种氮杂环咔唑类化合物(a1)的制备
[0093]
s1.将3,6-二叔丁基-9h-咔唑(1.12g,4.0mmol)、1-溴-4-碘苯、碘化亚铜、碳酸钾按1:1.2:0.2:2的摩尔比加入装有20ml无水n,n-二甲基甲酰胺的两颈烧瓶中,将烧瓶抽真空并在干燥氮气中置换三次,并在氮气、140℃下加热回流搅拌12h后,冷却,过滤,用乙酸乙酯萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚)得到1.15g白色粉末(中间体m1,产率为66%)。
[0094]
反应式如下所示:
[0095][0096]
s2.将步骤s1所得中间体m1(521mg,1.20mmol)、2,4-联苯-6-(4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)苯基)-1,3,5-三嗪、四(三苯基膦)钯、碳酸钾按1:1.05:0.047:2.51的摩尔比依次加入到带有回流冷凝器的25ml圆底烧瓶中,将烧瓶抽真空并在干燥氮气中置换三次后,再将20ml四氢呋喃和1.2ml去离子水注入烧瓶中,在氮气、80℃下回流12h,冷却,过滤,用二氯甲烷萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚/二氯甲烷=1:1(v/v))得到621mg白色粉末(式(a1)化合物,产率为78.07%)。反应式如下所示:
[0097][0098]
实施例2一种氮杂环咔唑类化合物(a2)的制备
[0099]
s1.将3,6-二叔丁基-9h-咔唑(279mg,1.2mmol)、5-溴-2-碘苯甲酸甲酯、碘化亚铜、碳酸钾按1:1.2:0.2:2的摩尔比加入装有5ml无水n,n-二甲基甲酰胺的两颈烧瓶中,将烧瓶抽真空并在干燥氩气中置换三次,并在氩气、140℃下加热回流搅拌12h后,冷却,过滤,用乙酸乙酯萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚)得到365mg白色粉末(中间体m2,产率为74%)。反应式如下所示:
[0100][0101]
s2.将苯基溴化镁在氩气下加入烧瓶中搅拌,再将溶解有中间体m2(365mg,0.74mmol)的1ml四氢呋喃缓慢加入烧瓶中,在50℃下加热4h,用0.1ml氢氧化铵水溶液淬灭并蒸发thf,加入二氯甲烷和水萃取,收集有机层并用na2so4干燥,产生的淡黄色粘性固体溶解于乙酸中,再逐滴加入36%盐酸,在120℃下回流4h后,冷却,加入二氯甲烷和水萃取,蒸发有机层后通过柱层析法(洗脱剂为石油醚)得到170mg白色粉末(中间体m3,产率为38.32%)。其中,所述中间体m2、苯基溴化镁、36%盐酸的摩尔比为5.03:1:2。反应式如下所示:
[0102][0103]
s3.将中间体m3(598mg,1.00mmol)、2,4-联苯-6-(4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)苯基)-1,3,5-三嗪、四(三苯基膦)钯、碳酸钾按1:1.05:0.047:2.51的摩尔比依次加入到带有回流冷凝器的25ml圆底烧瓶中,将烧瓶抽真空并在干燥氩气中置换三次后,再将6ml四氢呋喃和1.5ml去离子水注入烧瓶中,在氩气、80℃下回流12h,冷却,过滤,用二氯甲烷萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚/二氯甲烷=1:1(v/v))得到600mg白色粉末(式(a2)化合物,产率为72.55%)。反应式如下所示:
[0104][0105]
实施例3一种氮杂环咔唑类化合物(a1)的制备
[0106]
s1.将3,6-二叔丁基-9h-咔唑(1.12g,4.0mmol)、1-溴-4-碘苯、碘化亚铜、碳酸钾按1:1.2:0.3:3的摩尔比加入装有20ml无水二甲基亚砜的两颈烧瓶中,将烧瓶抽真空并在干燥氩气中置换三次,并在氩气、120℃下加热回流搅拌15h后,冷却,过滤,用乙酸乙酯萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚)得到1.06g白色粉末(中间体m1,产率为61%)。反应式同实施例1。
[0107]
s2.将步骤s1所得中间体m1(521mg,1.20mmol)、2,4-联苯-6-(4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)苯基)-1,3,5-三嗪、四(三苯基膦)钯、碳酸钾按1:1.2:0.06:2的摩尔比依次加入到带有回流冷凝器的25ml圆底烧瓶中,将烧瓶抽真空并在干燥氩气中置换三次后,再将20ml四氢呋喃和1.2ml去离子水注入烧瓶中,在氩气、70℃下回流14h,冷却,过滤,用二氯甲烷萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚/二氯甲烷=1:1(v/v))得到598mg白色粉末(式(a1)化合物,产率为75.21%)。反应式同实施例1。
[0108]
实施例4一种氮杂环咔唑类化合物(a1)的制备
[0109]
s1.将3,6-二叔丁基-9h-咔唑(1.12g,4.0mmol)、1-溴-4-碘苯、碘化亚铜、碳酸钠按1:1.5:0.2:2的摩尔比加入装有20ml无水n,n-二甲基乙酰胺的两颈烧瓶中,将烧瓶抽真空并在干燥氦气中置换三次,并在氦气、100℃下加热回流搅拌12h后,冷却,过滤,用乙酸乙酯萃取,脱溶剂后通过柱层析法(洗脱剂为石油醚)得到1.08g白色粉末(中间体m1,产率为
62%)。反应式同实施例1。
[0110]
s2.将步骤s1所得中间体m1(521mg,1.20mmol)、2,4-联苯-6-(4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)苯基)-1,3,5-三嗪、四(三苯基膦)钯、碳酸钠按1:1:0.04:3的摩尔比依次加入到带有回流冷凝器的25ml圆底烧瓶中,将烧瓶抽真空并在干燥氦气中置换三次后,再将20ml四氢呋喃和1.2ml去离子水注入烧瓶中,在氦气、85℃下回流10h,冷却,过滤,用二氯甲烷萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚/二氯甲烷=1:1(v/v))得到607mg白色粉末(式(a1)化合物,产率为76.25%)。反应式同实施例1。
[0111]
实施例5一种氮杂环咔唑类化合物(a2)的制备
[0112]
s1.将3,6-二叔丁基-9h-咔唑(279mg,1.0mmol)、5-溴-2-碘苯甲酸甲酯、碘化亚铜、碳酸钾按1:1.5:0.3:2的摩尔比加入装有5ml无水n,n-二甲基乙酰胺的两颈烧瓶中,将烧瓶抽真空并在干燥氮气中置换三次,并在氮气、120℃下加热回流搅拌15h后,冷却,过滤,用乙酸乙酯萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚)得到345mg白色粉末(中间体m2,产率为70%);
[0113]
s2.将苯基溴化镁在氮气下加入烧瓶中搅拌,再将溶解有中间体m2(365mg,0.74mmol)的1ml乙醚缓慢加入烧瓶中,在60℃下加热5h,用0.1ml氢氧化铵水溶液淬灭并蒸发thf,加入二氯甲烷和水萃取,收集有机层并用na2so4干燥,产生的淡黄色粘性固体溶解于乙酸中,再逐滴加入氢碘酸,在130℃下回流3h后,冷却,加入二氯甲烷和水萃取,蒸发有机层后通过柱层析法(洗脱剂为石油醚)得到156mg白色粉末(中间体m3,产率为35.26%)。其中,所述中间体m2、苯基溴化镁、氢碘酸的摩尔比为5:1:1.5。
[0114]
s3.将中间体m3(521mg,1.20mmol)、2,4-联苯-6-(4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)苯基)-1,3,5-三嗪、四(三苯基膦)钯、碳酸钾按1:1.2:0.06:2的摩尔比依次加入到带有回流冷凝器的25ml圆底烧瓶中,将烧瓶抽真空并在干燥氮气中置换三次后,再将6ml四氢呋喃和1.5ml去离子水注入烧瓶中,在氮气、70℃下回流14h,冷却,过滤,用二氯甲烷萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚/二氯甲烷=1:1(v/v))得到580mg白色粉末(式(a2)化合物,产率为70.15%)。
[0115]
实施例6一种氮杂环咔唑类化合物(a2)的制备
[0116]
s1.将3,6-二叔丁基-9h-咔唑(279mg,1.0mmol)、5-溴-2-碘苯甲酸甲酯、碘化亚铜、碳酸钠按1:1.2:0.2:3的摩尔比加入装有5ml无水二甲基亚砜的两颈烧瓶中,将烧瓶抽真空并在干燥氦气中置换三次,并在氦气、100℃下加热回流搅拌12h后,冷却,过滤,用乙酸乙酯萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚)得到360mg白色粉末(中间体m2,产率为73%);
[0117]
s2.将苯基溴化镁在氦气下加入烧瓶中搅拌,再将溶解有中间体m2(365mg,0.74mmol)的1ml甲苯缓慢加入烧瓶中,在50℃下加热4h,用0.1ml氢氧化铵水溶液淬灭并蒸发thf,加入二氯甲烷和水萃取,收集有机层并用na2so4干燥,产生的淡黄色粘性固体溶解于乙酸中,再逐滴加入36%盐酸,在110℃下回流5h后,冷却,加入二氯甲烷和水萃取,蒸发有机层后通过柱层析法(洗脱剂为石油醚)得到152mg白色粉末(中间体m3,产率为34.20%)。其中,所述中间体m2、苯基溴化镁、36%盐酸的摩尔比为6:1:3。
[0118]
s3.将中间体m3(521mg,1.20mmol)、2,4-联苯-6-(4-(4,4,5,5-四甲基-1,3,2-二
恶硼烷-2-基)苯基)-1,3,5-三嗪、四(三苯基膦)钯、碳酸钾按1:1:0.04:3的摩尔比依次加入到带有回流冷凝器的25ml圆底烧瓶中,将烧瓶抽真空并在干燥氦气中置换三次后,再将6ml四氢呋喃和1.5ml去离子水注入烧瓶中,在氦气、85℃下回流10h,冷却,过滤,用二氯甲烷萃取,分离出有机层并用无水硫酸镁干燥,过滤并蒸发后,粗产物通过柱层析法(洗脱剂为石油醚/二氯甲烷=1:1(v/v))得到590mg白色粉末(式(a2)化合物,产率为71.34%)。
[0119]
实验例结构表征及性能测试
[0120]
(1)核磁共振氢谱:
[0121]
采用布鲁克400mhz超导核磁共振仪对实施例1和2得到的氮杂环咔唑类化合物(a1和a2)进行核磁共振扫描,得到图1~2的核磁共振氢谱图。
[0122]
从图1可知,1h nmr(400mhz,cdcl3)δ=8.89(d,j=8.4,2h),8.81(dd,j=7.9,1.5,4h),8.17(d,j=1.4,2h),7.90(dd,j=8.3,6.7,4h),7.69(d,j=8.4,1h),7.64
–
7.57(m,3h),7.51
–
7.43(m,2h),1.47(d,j=10.2,9h)。分子氢谱波峰能与目标产物一一对应,数量合理。
[0123]
从图2可知,1h nmr(400mhz,cdcl3)δ=8.80
–
8.75(m,6h),8.16(dd,j=11.8,5.2,2h),7.99(dd,j=20.7,5.1,2h),7.72(dd,j=8.5,2.1,1h),7.63
–
7.54(m,9h),7.44(d,j=2.1,1h),7.27(dd,j=4.8,2.9,1h),7.25
–
7.20(m,5h),7.17
–
7.09(m,5h),1.48(s,9h),1.38(s,9h)。分子氢谱波峰能与目标产物一一对应,数量合理。
[0124]
(2)质谱:
[0125]
首先将含芴的菲并咪唑衍生物(5mg)溶于二氯甲烷中,再滴加乙腈至5ml,用0.22μm滤膜过滤,过滤除去超0.22um的微粒,使检测干扰降到最小,再分别将实施例1和2得到的氮杂环咔唑类化合物放入液相质谱联用仪中,利用试样中各组分电离生成不同荷质比的离子,经加速电场的作用,形成离子束,进入质量分析器,利用电场和磁场使发生相反的速度色散——离子束中速度较慢的离子通过电场后偏转大,速度快的偏转小;在磁场中离子发生角速度矢量相反的偏转,即速度慢的离子依然偏转大,速度快的偏转小;当两个场的偏转作用彼此补偿时,它们的轨道便相交于一点。与此同时,在磁场中还能发生质量的分离,使具有同一质荷比而速度不同的离子聚焦在同一点上,不同质荷比的离子聚焦在不同的点上,将它们分别聚焦而得到图3~4的质谱图,从而确定实施例1和2得到的氮杂环咔唑类化合物的质量。
[0126]
图3显示,实施例1得到的氮杂环咔唑类化合物的相对分子质量为663.35,与目标产物(a1)的相对分子质量一致。
[0127]
图4显示,实施例2得到的氮杂环咔唑类化合物的相对分子质量为827.47,与目标产物(a2)的相对分子质量一致。
[0128]
因此,基于核磁共振和质谱的结果,可以确定实施例1~2制备所得化合物的结构式分别如下式(a1)、式(a2)所示:
[0129][0130]
(3)电化学研究:
[0131]
使用循环伏安法对实施例1和2得到的氮杂环咔唑类化合物(a1和a2)进行测试,其氧化电位图如图5和6所示,根据参比电极为ag
+
/ag电极,电解质为0.1mol/l的agno3水溶液,由公式e(ag
+
/ag)=e
θ
(ag
+
/ag)+0.05916lgαag
+
可得ag
+
/ag电极电位:e(ag
+
/ag)=0.732v,而根据二茂铁在乙氰中参比于标准氢电极的氧化电位:0.630v有二茂铁在此ag
+
/ag参比电极下的氧化电位为:e
ox
(fc
+
/fc)-e(ag
+
/ag)=-0.102v。
[0132]
因此,实施例1所得氮杂环咔唑类化合物(a1)相对于二茂铁的电位差为:e
ox
(氮杂环咔唑类化合物(a1))-[e
ox
(fc
+
/fc)-e(ag
+
/ag)]=0.788v,而二茂铁相对于真空能级为4.8ev,故氮杂环咔唑类化合物(a1)的homo为:homo=-(ee
ox
(氮杂环咔唑类化合物(a1))-e[e
ox
(fc
+
/fc)-e(ag
+
/ag)]+4.8)=-5.55ev。利用紫外测定结果有,能隙eg为3.07ev,故lumo=homo+eg=-2.48ev。
[0133]
同理,实施例2所得氮杂环咔唑类化合物(a2)的homo为-5.49ev,lumo为-2.58ev。
[0134]
表明本发明的氮杂环咔唑类化合物(a1和a2)的lumo能级与经典的电子传输材料(tpbi)的lumo能级(-2.70ev)相接近,说明其载流子传输性能良好,即具有较高的导电率,能较好地传输电子,适用于制备成发光器件。
[0135]
(4)热重分析:
[0136]
采用高温同步热分析仪对实施例1和2的氮杂环咔唑类化合物(a1和a2)分别进行热重分析,得到图7的热重分析图。测定条件:在氮气保护下,升温速率为10℃/min,测量温度范围为30~800℃。
[0137]
图7显示,该氮杂环咔唑类化合物(a1和a2)分别表现出高达400℃和420℃的热分解温度(td),说明本发明的氮杂环咔唑类化合物能在较高的温度下相对稳定,具有较优的热稳定性,为真空蒸镀工艺制作器件提供了必要的条件。
[0138]
(5)相转变温度测试:
[0139]
采用低温差示扫描量热仪对实施例1和2的氮杂环咔唑类化合物(a1和a2)分别进行相转变温度测试,结果如图8所示,可见该氮杂环咔唑类化合物(a1和a2)的玻璃化转变温度分别为120℃和150℃,说明本发明的氮杂环咔唑类化合物具有较优的成膜性,可适用于制备成为发光材料。
[0140]
(6)溶剂化效应测试:
[0141]
仪器:爱丁堡fl980瞬态稳态荧光磷光光谱仪;测试方法:参数设置。设置激发波长
375nm,设置狭缝宽度使其纵坐标数值接近一百万,再进行光谱测试获得图9和10的溶剂化效应图。
[0142]
在7种不同极性的溶剂(按极性从小到大排序,为:正己烷<甲苯<氯仿<<二氯甲烷<四氢呋喃<乙酸乙酯<乙腈)中测量了实施例1所得氮杂环咔唑类化合物(a1)在荧光方面的溶剂化效应,其结果如图9所示:1)在低极性的溶剂如正己烷中,a1的荧光光谱表现出由局部激发态引起的较佳的振动结构;2)随着溶剂极性的增加,荧光光谱不断变宽并逐渐变得无结构;3)提高溶剂的极性共产生188nm的红移(正己烷的398nm到乙腈的586nm)。可见,该氮杂环咔唑类化合物(a1)的发射是局域激发与电荷转移的混合,具有较优的发射亮度。
[0143]
而实施例2所得氮杂环咔唑类化合物(a2)也表现出相同的现象(图10):提高溶剂的极性共产生145nm的红移(正己烷的410nm到乙腈的550nm),表明该氮杂环咔唑类化合物(a2)具有较优的发射亮度。
[0144]
(7)荧光量子产率测试:
[0145]
仪器:爱丁堡fl980瞬态稳态荧光磷光光谱仪;测试方法:参数设置,用产物的最佳激发波长375nm激发,硫酸奎宁做参比,保持激发与发射的狭缝宽度一致,结果测得实施例1和2的氮杂环咔唑类化合物(a1和a2)的荧光量子产率分别为80.78%和65.14%,表明本发明的氮杂环咔唑类化合物具有较优的荧光量子产率。
[0146]
(8)荧光光谱:
[0147]
图11~12为实施例1~2所得产物(a1和a2)在25℃下,甲苯溶液中分别鼓入氮气和氧气后荧光强度变化的荧光光谱图。图11~12显示,a1和a2在氮气气氛下的荧光发射强度比氧气气氛下的更高,说明氧气会使本发明的氮杂环咔唑类化合物(a1和a2)发生三重态的荧光猝灭,使荧光发射强度有所降低,表明本发明的氮杂环咔唑类化合物(a1和a2)能利用来自三重态的激子,提高激子的利用率,实现较优的荧光量子产率。
[0148]
(9)薄膜荧光发射:
[0149]
利用涂膜机将实施例1~2所得产物(a1和a2)分别以10wt%的用量掺杂于主体dpepo中旋涂成薄膜,再分别利用爱丁堡fl980瞬态稳态荧光磷光光谱仪进行测试,得到图13的荧光发射光谱。从图中可以看到a1和a2的最大发射波长分别为450nm和470nm,均处于深蓝光的区域里,因此a1和a2可适用于制备深蓝光材料。
[0150]
综上,本发明的氮杂环咔唑类化合物具有可作为深蓝光材料来源的卓越性能,如具有良好的热稳定性、较优的成膜性、较强的发射亮度、较高的荧光量子产率、较佳的传输电子能力,作为一种成本低、性能佳的新型有机发光小分子,十分适合制备成发光材料或发光器件。
[0151]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。