1.本发明属于有机化学合成技术领域,具体涉及一种3-取代二苯并噻吩及其合成方法。
背景技术:
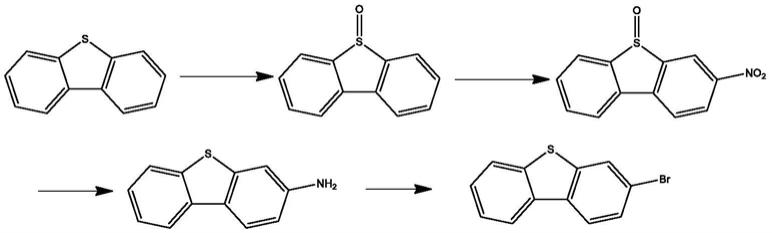
2.二苯并噻吩类的有机化合物在有机发光器件中有广泛的应用,而3-取代二苯并噻吩是一类重要的有机分子骨架,现有技术的合成中3-卤代二苯并噻吩工艺成本较高,反应过程生产很多废水,污染较大,危险性较大,对设备的要求较高,后处理难度大,以下参考已有合成路线来阐述现有工艺的缺点。
3.目前此类化合物的合成方法主要有以下几种:
4.常规路线1(参照jp2018090561,wo2017196081a1,wo2002078693)
[0005][0006]
常规路线2(参照cn112552279a,文献organic letters,2018,vol.20,#17,p.5439-5443)
[0007][0008]
常规路线3(参照文献organic letters,2016,vol.18,#21,p.5756-5759)
[0009][0010]
针对以上三种合成方法分析可知,现有的合成方法存在很多问题,例如常规路线1中使用间氯过氧苯甲酸或者双氧水进行第一步的氧化,氧化得到的产物亚砜会进一步被氧化成砜,此杂质较难控制且在后续会很难除去,就导致了最终产物纯度很难得到较高的纯度;另间氯过氧苯甲酸(常规路线3中用于制备碘嗡盐)在放大时会有安全的风险。在第二步反应中使用浓硫酸,硝酸和醋酸进行硝化反应,会产生大量的酸性废液,造成环境的污染。第三步使用加氢釜还原对于设备要求较高,或者使用氯化亚锡和盐酸还原,反应产生一些不易除去的无机物质,造成后处理较难,仍然无法放大操作。常规路线1第四步反应,常规路线2第二步反应和常规路线3第一步反应,均需要制备重氮盐溶液,该过程存在一定的危险性,对于放大生产设备要求同样很高,不适合大量生产,且产生掉氨基的杂质,使产品得到高纯度产品难度增大。常规路线2中使用正丁基锂和乙二硫醇制备反应中,选择性较差,容易产生很多杂质,导致最终产品纯化难度大,且影响收率,另乙二硫醇臭味较大,不适合放大生产。另常规路线2第二步反应和常规路线3中使用dmso,后处理时大量的dmso较难水洗除去,导致产品溶解不易析出,影响产品收率;且常规路线3中使用碳酸铯用量较大,成本较高。因此,研发一种新的取代二苯并噻吩的合成方法对实现工业化生产具有重要的意义。
技术实现要素:
[0011]
本发明的目的在于克服上述现有技术的不足:1、对设备要求高,后处理复杂;2、反应过程产生大量废酸及废水,污染环境;3、重氮盐反应,操作具有一定的危险性;4、产生杂质较多,不能得到高纯度的最终产品;5、不适合工艺放大批量生产操作。
[0012]
为了解决技术问题,本发明的技术方案是:一种3-取代二苯并噻吩的合成方法,包括以下步骤:
[0013]
步骤1:以邻碘茴香硫醚和4-取代苯硼酸为起始原料,进行偶联反应,得到中间体a,所述邻碘茴香硫醚与4-取代苯硼酸的摩尔比为1:1~1.4;
[0014]
步骤2:中间体a、对甲苯磺酸和双氧水进行氧化反应,得到中间体b,所述中间体a、对甲苯磺酸和双氧水的摩尔比为1:0.5:1~1.2;
[0015]
步骤3:中间体b和伊顿试剂进行关环反应,得到3-取代二苯并噻吩,所述中间体b与伊顿试剂的摩尔比1:1~1.5;所述3-取代二苯并噻吩的结构式为
其中r为br、f、cl、cf3、oh、benzene或no2。
[0016]
优选的,所述步骤1具体为:在氮气保护下,向反应瓶中加入邻碘茴香硫醚、4-取代苯硼酸、甲苯、乙醇、水和碳酸钾,加完后搅拌,加热升温至60~70℃,迅速加入四(三苯基膦)钯,继续升温至75~80℃,回流反应10~12h,反应完毕后降温,得到中间体a反应体系;所述邻碘茴香硫醚与4-取代苯硼酸的摩尔比为1:1.2,邻碘茴香硫醚、甲苯、乙醇和水的用量比为1kg:4l:2l:2l,邻碘茴香硫醚与碳酸钾的摩尔比为1:2,邻碘茴香硫醚与四(三苯基膦)钯的摩尔比为10:0.1。
[0017]
优选的,所述中间体a反应体系利用甲苯进行萃取,有机相水洗,有机相干燥,过滤并除去干燥剂,浓缩有机相,浓缩物使用甲醇重结晶,烘干得到中间体a。
[0018]
优选的,所述4-取代苯硼酸的结构式为其中r为br、f、cl、cf3、oh、benzene、no2或h。
[0019]
优选的,所述r为h时,4-取代苯硼酸为苯硼酸,邻碘茴香硫醚和苯硼酸进行偶联反应,得到中间体a,中间体a、对甲苯磺酸和双氧水进行氧化反应,得到中间体c向三口瓶中加入中间体c、乙酸和二氯乙烷,搅拌溶解澄清,温度升至25℃,开始滴加溴素,体系放热至40℃,控制温度于25℃~50℃,滴加完毕,在此温度下反应约20h,反应完毕,使用亚硫酸氢钠水溶液洗涤,有机相干燥,过滤并除去干燥剂,浓缩有机相,得到中间体b,所述中间体c与溴素的摩尔比为1:1,中间体c与乙酸和二氯乙烷的用量比为1kg:2l:2l。
[0020]
优选的,所述步骤2具体为:向反应瓶中加入步骤1制备得到的中间体a和对甲苯磺酸,加入二氯乙烷溶解澄清,在20~30℃下,加入双氧水,室温20~30℃反应3~5h,停止反应后得到中间体b反应体系,所述中间体a、对甲苯磺酸和双氧水的摩尔比为2:1:2,中间体a与二氯乙烷的用量比为1kg:4l,其中双氧水的浓度为30%。
[0021]
优选的,所述中间体b反应体系利用二氯乙烷进行萃取,有机相水洗,有机相干燥,过滤并除去干燥剂,浓缩有机相,使用石油醚进行分散,得到中间体b。
[0022]
优选的,所述步骤3具体为:向反应瓶中加入步骤2得到的中间体b、二氯乙烷和伊顿试剂,开启搅拌和加热,升温至60℃~80℃,在此温度下反应6~8h,反应完全后得到3-取代二苯并噻吩反应体系,中间体b与二氯乙烷的用量比为1kg:2l,中间体b与伊顿试剂的摩尔比1:1。
[0023]
优选的,所述3-取代二苯并噻吩反应体系在搅拌下倒入冰水中,使用三乙胺进行ph调节,ph调节至7~8之间,过滤析出固体,并使用乙醇进行淋洗,抽干并干燥得到3-取代二苯并噻吩。
[0024]
优选的,一种3-取代二苯并噻吩,利用上述所述的3-取代二苯并噻吩的合成方法进行合成,3-取代二苯并噻吩的结构式为:所述r为br、f、cl、cf3、oh、benzene或no2。
[0025]
与现有技术相比,本发明的优点在于:
[0026]
(1)本发明第一步反应采用邻碘茴香硫醚和4-取代苯硼酸经过suzzki反应,高收率得到偶联产物,副产物极少,且原料成本较低,反应对设备要求低;
[0027]
(2)本发明第二步反应在对甲苯磺酸和双氧水(30%)条件下进行氧化反应,氧化反应在室温条件下进行,反应条件较温和,反应产物单一,后处理简单;
[0028]
(3)本发明第三步反应使用二氯乙烷或者甲苯做溶剂,加入当量的伊顿试剂进行关环反应,反应时间较短,大大减少了废酸的产生,环境友好,并使用三乙胺进行中和至中性或者弱碱性后直接过滤的方式,简化了后处理方式,较容易得到高纯度的产品;
[0029]
(4)本发明合成的3-取代二苯并噻吩纯度较高,后处理较简单,污染较小,适合于工艺大批量的生产;本发明所用的原材料、溶剂和催化剂均为常规市售产品。
附图说明
[0030]
图1、本发明实施例1和实施例2产物的1h nmr表征谱图;
[0031]
图2、本发明实施例3产物的1h nmr表征谱图;
[0032]
图3、本发明实施例4产物的1h nmr表征谱图;
[0033]
图4、本发明实施例5产物的1h nmr表征谱图;
[0034]
图5、本发明实施例6产物的1h nmr表征谱图;
[0035]
图6、本发明实施例7产物的1h nmr表征谱图;
[0036]
图7、本发明实施例8产物的1h nmr表征谱图。
具体实施方式
[0037]
以下结合具体实施例对本发明进行说明,所用的原材料、溶剂和催化剂均为常规市售产品,以下实施例用于说明本发明,但不用来限制本发明的范围。
[0038]
本发明公开了一种3-取代二苯并噻吩的合成方法,包括以下步骤:
[0039]
步骤1:以邻碘茴香硫醚和4-取代苯硼酸为起始原料,进行偶联反应,得到中间体a,所述邻碘茴香硫醚与4-取代苯硼酸的摩尔比为1:1~1.4;
[0040]
步骤2:中间体a、对甲苯磺酸和双氧水进行氧化反应,得到中间体b,所述中间体a、对甲苯磺酸和双氧水的摩尔比为1:0.5:1~1.2;
[0041]
步骤3:中间体b和伊顿试剂进行关环反应,得到3-取代二苯并噻吩,所述中间体b
与伊顿试剂的摩尔比1:1~1.5;所述3-取代二苯并噻吩的结构式为其中r为br、f、cl、cf3、oh、benzene或no2。
[0042]
所述伊顿试剂为7.7wt%的五氧化二磷的甲磺酸溶液,此试剂可以代替多聚磷酸用于催化酰基化反应,最常用是关环反应(五元或六元环)。
[0043][0044]
所述邻碘茴香硫醚也可用邻碘茴香硫醚的衍生物进行替代。
[0045]
优选的,所述步骤1具体为:在氮气保护下,向反应瓶中加入邻碘茴香硫醚、4-取代苯硼酸、甲苯、乙醇、水和碳酸钾,加完后搅拌,加热升温至60~70℃,迅速加入四(三苯基膦)钯,继续升温至75~80℃,回流反应10~12h,反应完毕后降温,得到中间体a反应体系;所述邻碘茴香硫醚与4-取代苯硼酸的摩尔比为1:1.2,邻碘茴香硫醚、甲苯、乙醇和水的用量比为1kg:4l:2l:2l,邻碘茴香硫醚与碳酸钾的摩尔比为1:2,邻碘茴香硫醚与四(三苯基膦)钯的摩尔比为10:0.1。
[0046]
优选的,所述中间体a反应体系利用甲苯进行萃取,有机相水洗,有机相干燥,过滤并除去干燥剂,浓缩有机相,浓缩物使用甲醇重结晶,烘干得到中间体a。
[0047]
优选的,所述4-取代苯硼酸的结构式为其中r为br、f、cl、cf3、oh、benzene、no2或h。
[0048]
优选的,所述r为h时,4-取代苯硼酸为苯硼酸,邻碘茴香硫醚和苯硼酸进行偶联反应,得到中间体a,中间体a、对甲苯磺酸和双氧水进行氧化反应,得到中间体c向三口瓶中加入中间体c、乙酸和二氯乙烷,搅拌溶解澄清,温度升至25℃,开始滴加溴素,体系放热至40℃,控制温度于25℃~50℃,滴加完毕,在此温度下反应约20h,反应完毕,使用亚硫酸氢钠水溶液洗涤,有机相干燥,过滤并除去干燥剂,浓缩有机相,得到中间体b,所述中间体c与溴素的摩尔比为1:1,中间体c与乙酸和二氯乙烷的用量比为1kg:2l:2l。
[0049]
优选的,所述步骤2具体为:向反应瓶中加入步骤1制备得到的中间体a和对甲苯磺
酸,加入二氯乙烷溶解澄清,在20~30℃下,加入双氧水,室温20~30℃反应3~5h,停止反应后得到中间体b反应体系,所述中间体a、对甲苯磺酸和双氧水的摩尔比为2:1:2,中间体a与二氯乙烷的用量比为1kg:4l,其中双氧水的浓度为30%。
[0050]
优选的,所述中间体b反应体系利用二氯乙烷进行萃取,有机相水洗,有机相干燥,过滤并除去干燥剂,浓缩有机相,使用石油醚进行分散,得到中间体b。
[0051]
优选的,所述步骤3具体为:向反应瓶中加入步骤2得到的中间体b、二氯乙烷和伊顿试剂,开启搅拌和加热,升温至60℃~80℃,在此温度下反应6~8h,反应完全后得到3-取代二苯并噻吩反应体系,中间体b与二氯乙烷的用量比为1kg:2l,中间体b与伊顿试剂的摩尔比1:1。
[0052]
优选的,所述3-取代二苯并噻吩反应体系在搅拌下倒入冰水中,使用三乙胺进行ph调节,ph调节至7~8之间,过滤析出固体,并使用乙醇进行淋洗,抽干并干燥得到3-取代二苯并噻吩。
[0053]
优选的,一种3-取代二苯并噻吩,利用上述所述的3-取代二苯并噻吩的合成方法进行合成,3-取代二苯并噻吩的结构式为:所述r为br、f、cl、cf3、oh、benzene或no2。
[0054]
实施例1:
[0055][0056]
其中,为邻碘茴香硫醚,为苯硼酸。
[0057]
步骤1:在氮气保护下,向反应瓶中加入邻碘茴香硫醚(2.50kg,10mol)、苯硼酸(1.46kg,12mol)、甲苯10l、乙醇5l、水5l,碳酸钾(2.76kg,20mol),加完,搅拌,加热升温至60~70℃,迅速加入四(三苯基膦)钯(115g,0.1mol),继续升温至75~80℃回流反应10h,反应完毕,降温,用甲苯萃取反应体系,有机相水洗,有机相干燥,过滤并除去干燥剂,浓缩有机相,浓缩物使用甲醇重结晶至hplc>98.5%,烘干得到中间体a-1,收率90.2%。
[0058]
步骤2:向反应瓶中加入上步制备得到的中间体a-1(1.80kg,9mol)、对甲苯磺酸(0.856g,4.5mol),加入二氯乙烷7.2l溶解澄清,在20~30℃下,加入30%h2o2(1.02kg,9mol),室温(20~30℃)反应5h,停止反应后,二氯乙烷萃取反应体系,有机相水洗,有机相干燥,过滤并除去干燥剂,浓缩有机相,使用石油醚进行分散,得到中间体c-1,hplc>98.5%,收率97.1%。
[0059]
步骤3:向三口瓶中加入上步制备得到的中间体c-1(1.30kg,6mol),乙酸2.6l,二氯乙烷2.6l,搅拌溶解澄清,温度升至25℃,开始滴加溴素(0.96kg,6mol),体系放热至40℃,控制温度于25℃~50℃,滴加完毕,在此温度下反应约20h,反应完毕,使用亚硫酸氢钠水溶液洗涤,有机相干燥,过滤并除去干燥剂,浓缩有机相,得到中间体b-1,hplc>98.5%,收率98.5%。
[0060]
步骤4:向反应瓶中加入上步制备得到的中间体b-1(1.48kg,5mol),二氯乙烷3l,伊顿试剂(1.20kg,5mol),开启搅拌和加热,升温至60℃~80℃,在此温度下反应6h,反应完全后,反应液在搅拌下倒入冰水中,使用三乙胺进行调节ph=7~8之间,过滤析出固体,并使用乙醇进行淋洗,抽干并干燥得到3-溴二苯并噻吩,hplc>99.5%,收率90.3%。
[0061]
实施例2:
[0062][0063]
其中,为邻碘茴香硫醚,为对溴苯硼酸。
[0064]
步骤1:在氮气保护下,向反应瓶中加入邻碘茴香硫醚(2.5kg,10mol)、对溴苯硼酸(1.46kg,12mol)、甲苯10l、乙醇5l、水5l,碳酸钾(2.76kg,20mol),加完,搅拌,加热升温至60~70℃,迅速加入四(三苯基膦)钯(115g,1mol),继续升温至75~80℃回流反应10h,反应完毕,降温,用甲苯萃取反应体系,有机相水洗,有机相干燥,过滤并除去干燥剂,浓缩有机相,浓缩物使用甲醇重结晶至hplc>97.8%,烘干得到中间体a-2,收率65.5%。
[0065]
步骤2:向反应瓶中加入上步制备得到的中间体a-2(1.30kg,6mol)、对甲苯磺酸(0.570kg,3mol),加入二氯乙烷5.4l溶解澄清,在20~30℃下,加入30%h2o2(0.68kg,6mol),室温(20~30℃)反应5h,停止反应后,二氯乙烷萃取反应体系,有机相水洗,有机相干燥,过滤并除去干燥剂,浓缩有机相,使用石油醚进行分散,得到中间体b-2,hplc>98.5%,收率98.5%。
[0066]
步骤3:向反应瓶中加入上步制备得到的中间体b-2(1.48kg,5mol),二氯乙烷3l,伊顿试剂(1.20kg,5mol),开启搅拌和加热,升温至60℃~80℃,在此温度下反应6h,反应完全后,反应液在搅拌下倒入冰水中,使用三乙胺进行调节ph=7~8之间,过滤析出固体,并使用乙醇进行淋洗,抽干并干燥得到3-溴二苯并噻吩,hplc>98.0%,收率92.1%。
[0067]
如图1所示,实施例1和2合成的最终产品均经过1h nmr表征,1h nmr(400mhz,cdcl3):δ8.14-8.07(m,1h),8.00-7.95(m,2h),7.86-7.81(m,1h),7.55(dd,1h),7.51-7.43(m,2h),该核磁谱图数据与产物结构吻合。
[0068]
实施例3~8:
[0069][0070]
其中r=f,cl,cf3,oh,benzene,no2[0071][0072]
步骤1:除使用不同的4-取代苯硼酸外,其他反应条件相同,具体如下:
[0073]
在氮气保护下,向反应瓶中加入邻碘茴香硫醚(10mol)、4-取代苯硼酸(14mol或10mol)、甲苯10l,乙醇5l,水5l,碳酸钾(20mol),加完,搅拌,加热升温至60~70℃,迅速加入四(三苯基膦)钯(1mol),继续升温至75~80℃回流反应10~15h,反应完毕,降温,用甲苯萃取反应体系,有机相水洗,有机相干燥,过滤并除去干燥剂,浓缩有机相,浓缩物使用甲醇重结晶,烘干得到中间体a-3~a-8。
[0074]
步骤2:向反应瓶中加入上步制备得到的中间体a-3~a-8(8mol)、对甲苯磺酸(4mol),加入二氯乙烷7.2l溶解澄清,在20~30℃下,加入30%h2o2(9.6mol),室温(20~30℃)反应3~5h,停止反应后,二氯乙烷反应体系,有机相水洗,有机相干燥,过滤并除去干燥剂,浓缩有机相.使用石油醚进行分散,得到中间体b-3~b-8。
[0075]
步骤3:向反应瓶中加入上步制备得到的中间体b-3~b-8(5mol),二氯乙烷3l,伊顿试剂(7.5mol),开启搅拌和加热,升温至60℃~80℃,在此温度下反应6~10h,反应完全后,反应液在搅拌下倒入冰水中,使用三乙胺进行调节ph=7~8之间,过滤析出固体,并使用乙醇进行淋洗,抽干并干燥得到3-取代二苯并噻吩。
[0076]
实施例3~8中邻碘茴香硫醚与4-取代苯硼酸反应见表如下:
[0077]
[0078][0079]
如图2~7所示,实施例3~8合成的最终产品均经过1h nmr表征,谱图数据与结构吻合,具体为:
[0080]
实施例3:1h nmr(400mhz,cdcl3):δ8.08(dd,2h),7.87-7.80(m,1h),7.54(d,1h),7.50-7.41(m,2h),7.19(d,1h)。
[0081]
实施例4:1h nmr(400mhz,cdcl3):δ8.13-8.07(m,1h),8.04(d,1h),7.87-7.79(m,2h),7.52-7.39(m,3h)。
[0082]
实施例5:1h nmr:(400mhz,cdcl3):δ8.30-8.09(m,3h),7.90(d,1h),7.70(d,1h),7.59-7.46(m,2h)。
[0083]
实施例6:1h nmr(400mhz,cdcl3):δ5.52(s,1h),7.68(dd,1h),8.10-8.26(m,3h),8.50(d,1h),8.85(dd,1h),9.19(dd,1h)。
[0084]
实施例7:1h nmr(400mhz,cdcl3):δ8.54-8.40(m,1h),8.22(dd,1h),8.02-7.90(m,1h),7.83(d,1h),7.68-7.53(m,2h),7.36(d,1h)。
[0085]
实施例8:1h nmr(500mhz,cdcl3):δ8.24(d,1h),8.21-8.19(m,1h),8.10(d,1h),7.92-7.88(m,1h),7.74-7.72(m,3h),7.54-7.49(m,4h),7.42(t,1h)。
[0086]
本发明反应原理如下:
[0087][0088]
其中r=f,cl,br,cf3,oh,benzene,no2[0089]
本发明以邻碘茴香硫醚或其衍生物和4-取代苯硼酸为起始原料,进行偶联反应,得到中间体a,高收率得到偶联产物,副产物极少,且原料成本较低,接着中间体a、对甲苯磺酸和双氧水进行氧化反应,得到中间体b,氧化反应在室温条件下进行,反应条件较温和,反应产物单一,然后中间体b和伊顿试剂进行关环反应,得到3-取代二苯并噻吩,关环反应反应时间较短,大大减少了废酸的产生,并使用三乙胺进行中和至中性或者弱碱性后直接过滤的方式,简化了后处理方式,较容易的得到高纯度的产品,所述3-取代二苯并噻吩的结构式为其中r为f、cl、cf3、oh、benzene或no2。
[0090]
本发明第一步反应采用邻碘茴香硫醚和4-取代苯硼酸经过suzzki反应,高收率得到偶联产物,副产物极少,且原料成本较低,反应对设备要求低。
[0091]
本发明第二步反应在对甲苯磺酸和双氧水(30%)条件下进行氧化反应,氧化反应在室温条件下进行,反应条件较温和,反应产物单一,后处理简单。
[0092]
本发明第三步反应使用二氯乙烷或者甲苯做溶剂,加入当量的伊顿试剂进行关环反应,反应时间较短,大大减少了废酸的产生,环境友好,并使用三乙胺进行中和至中性或者弱碱性后直接过滤的方式,简化了后处理方式,较容易得到高纯度的产品。
[0093]
本发明合成的3-取代二苯并噻吩纯度较高,后处理较简单,污染较小,适合于工艺大批量的生产;本发明所用的原材料、溶剂和催化剂均为常规市售产品。
[0094]
上面对本发明优选实施方式作了详细说明,但是本发明不限于上述实施方式,在本领域普通技术人员所具备的知识范围内,还可以在不脱离本发明宗旨的前提下做出各种变化。
[0095]
不脱离本发明的构思和范围可以做出许多其他改变和改型。应当理解,本发明不限于特定的实施方式,本发明的范围由所附权利要求限定。