一种7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备方法
技术领域
1.本发明涉及药物化学领域,具体涉及一种7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备方法。
背景技术:
2.7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮是合成溴芬酸钠的重要中间体,溴芬酸钠由日本千寿制药株式会社开发为滴眼液上市,于2000年3月获得日本pmda批准上市,用于外眼部及前眼部的炎症性疾病对症治疗。
[0003][0004]
目前7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备方法主要包括以下两种:
[0005]
方法一,美国专利us 4126635和journal of medicinal chemistry,1990 33(8),2296-2304报道了以2-氨基-4'-溴二苯甲酮和2-甲巯基乙酸乙酯为原料,用特戊酰氯催化下环合,再经雷尼镍或锡还原,该路线成环反应条件需要-70℃,工艺较复杂。
[0006][0007]
方法二,欧洲专利ep0221753、journal of heterocyclic chemistry,17(8),1663-4,1980和journal of medicinal chemistry 27(11),1379-1388,1984报道了以对溴苯甲腈和吲哚啉为原料,用三氯化硼和三氯化铝为催化剂,进行houben-hoesch反应,再经活性二氧化锰氧化、nbs或n-氯代丁二酰亚胺卤化、磷酸酸水解制备。该路线中缺点是磷酸水解时反应温度高时间长,有红色聚合物导致分离纯化困难。
[0008][0009]
现有技术中,7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备方法存在着各种不同的缺陷,寻找一种反应条件温和,纯化操作简便,易于工业化生产的工艺路线显然格外重要。
技术实现要素:
[0010]
本发明的目的在于克服现有技术的不足,提供一种7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备方法,该方法具有原料易得,反应条件温和,操作简单,收率高,纯度高,适用于工业化生产的特点。
[0011]
本发明通过如下技术方案实现:
[0012]
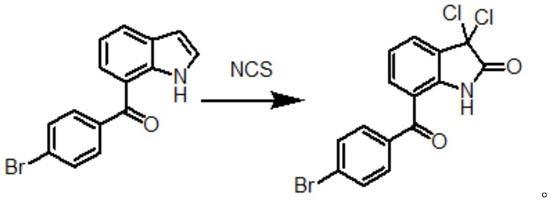
7-(4-溴苯甲酰基)吲哚进行卤代反应制得α-双卤代酰胺即3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮,3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮进行还原反应制得7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮。
[0013][0014]
其中,r为卤素,选自f、cl、br、i。
[0015]
进一步地,本发明包括如下步骤:
[0016]
(1)7-(4-溴苯甲酰基)吲哚在酸存在下,在四氢呋喃中卤代反应制得3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮;
[0017]
(2)3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮,在四氢呋喃中,在醋酸和锌粉作用下进行还原反应制得7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮。
[0018]
步骤(1)中,所述的酸为硫酸或盐酸,优选为浓硫酸或浓盐酸。
[0019]
步骤(1)中,所述的卤代反应的卤代试剂为n-氯代丁二酰亚胺、n-溴代丁二酰亚胺、或n-碘代丁二酰亚胺。
[0020]
优选地卤代试剂为n-氯代丁二酰亚胺。
[0021]
步骤(1)中,7-(4-溴苯甲酰基)吲哚:卤代试剂:酸:水:四氢呋喃的质量比为1:0.5-2.5:0.1-3:0-5:7-20;
[0022]
进一步地,为了保障反应的安全,本发明优选7-(4-溴苯甲酰基)吲哚:卤代试剂:酸:水:四氢呋喃的质量比为1:1.4-2:0.2-1.0:0.5-2:9-11;
[0023]
步骤(2)中,3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮:醋酸:锌粉的摩尔比为1:1-4:2-4。
[0024]
优选的反应流程如下:
[0025][0026]
本发明还涉及化合物3,3-二氯代-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮及其制备方法。
[0027]
所述的3,3-二氯代-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮结构如下:
[0028][0029]
其制备方法如下:
[0030]
7-(4-溴苯甲酰基)吲哚在酸存在下,在四氢呋喃中氯代反应制得3,3-二氯代-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮:
[0031][0032]
其中,所述的酸为硫酸或盐酸,优选为浓硫酸或浓盐酸。
[0033]
所述的氯化反应的氯代试剂为n-氯代丁二酰亚胺。
[0034]
7-(4-溴苯甲酰基)吲哚:氯代试剂:酸:水:四氢呋喃的质量比为1:0.5-2.5:0.1-3:0-5:7-20;
[0035]
优选地,7-(4-溴苯甲酰基)吲哚:卤代试剂:酸:水:四氢呋喃的质量比为1:1.4-2:0.2-1.0:0.5-2:9-11。
[0036]
本发明提供了7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备方法,以3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮为关键中间体合成7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮新的制备方法,该方法具有原料易得,反应条件温和可控,操作简单,适合工业化生产,同时提供了新化合物3,3-二氯代-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮及其制备方法,且以3,3-二氯代-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮作
为中间体制备7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮时,不仅成本低,而且收率高,操作简单,更适合工业化生产。
附图说明
[0037]
图1为3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的hplc图谱;
[0038]
图2为3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的氢谱;
[0039]
图3为3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的质谱;
[0040]
图4为7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的hplc图谱;
[0041]
图5为7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的氢谱;
[0042]
图6为7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的质谱。
具体实施方式
[0043]
下面将通过实施例对本发明作进一步的描述,这些描述并不是对本发明内容作进一步限定,对本发明的技术特征所做的等同替换或相应的改进,仍属于本发明的保护范围之内。
[0044]
实施例1 3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备:
[0045]
称取四氢呋喃9kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸0.1kg,水0.1kg,加入n-氯代丁二酰亚胺1.5kg,反应4小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体1.16kg,收率90%。1h-nmr(dmso-d6,600mhz)7.28(t,1h,j=7.8hz),7.56(dd,1h,j=7.9hz),7.71(d,2h,j=6.7hz),7.80(d,2h,j=6.7hz),7.96(d,1h,j=7.4hz),11.41(s,1h)。hrms(esi,neg)[m-h]=384。
[0046]
实施例2 3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备:
[0047]
称取四氢呋喃9kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸0.5kg,加入n-氯代丁二酰亚胺1.0kg,反应4小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体0.9kg,收率70%。
[0048]
实施例3 3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备:
[0049]
称取四氢呋喃10kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸1.0kg,水2.0kg,加入n-氯代丁二酰亚胺2.0kg,反应4小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体1.2kg,收率94%。
[0050]
实施例4 3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备:
[0051]
称取四氢呋喃7kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸0.2kg,加入n-氯代丁二酰亚胺0.5kg,反应4小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体1.15kg,收率70%。
[0052]
实施例5 3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备:
[0053]
称取四氢呋喃10kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸3.0kg、水5.0kg,加入n-氯代丁二酰亚胺1.4kg,反应4小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体1.21kg,收率95%。
[0054]
实施例6 3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备:
[0055]
称取四氢呋喃20kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸0.6kg、水1.0kg,
加入n-氯代丁二酰亚胺1.5kg,反应4小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体1.2kg,收率94%。
[0056]
实施例7 3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备:
[0057]
称取四氢呋喃10kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸1.0kg,加入n-氯代丁二酰亚胺1.4kg,反应4小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体1.2kg,收率94%。
[0058]
实施例8 3,3-二溴-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备:
[0059]
称取四氢呋喃9kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸0.2kg、水0.5kg,加入n-溴代丁二酰亚胺1.8kg,反应5小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体1.46kg,收率93%。
[0060]
实施例9 3,3-二碘-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备:
[0061]
称取四氢呋喃12kg、7-(4-溴苯甲酰基)吲哚1.0kg和36-38%盐酸0.5kg、水0.5kg,加入n-碘代丁二酰亚胺2.25kg,反应6小时后,加纯化水析晶抽滤,滤饼50℃干燥得浅黄固体1.8kg,收率95%。
[0062]
实施例10 7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备:
[0063]
实施例1制备的3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮1kg,四氢呋喃15kg和醋酸0.5kg(3.2equiv.)溶解,搅拌下加入锌粉0.675kg(4equv.),40℃反应2小时滤除不溶物后,所得滤液浓缩回收溶剂后加乙酸乙酯打浆得类白色固体0.6kg,收率73%。1h-nmr(dmso-d6,600mhz)3.59(s,2h),7.05(t,1h,j=7.5hz),7.33(d,1h,j=8hz),7.48(d,1h,j=7.3hz),7.66(d,2h,j=6.7hz),7.78(d,2h,j=6.7hz),10.36(s,1h)。hrms(esi,neg)[m+h]=316,318。
[0064]
实施例11 7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备:
[0065]
实施例1制备的3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮1kg,四氢呋喃13kg和醋酸0.156(1equiv.)kg溶解,搅拌下加入锌粉0.7kg(4.1equiv.),35℃反应2小时滤除不溶物后,所得滤液浓缩回收溶剂后加乙醇打浆得类白色固体0.66kg,收率80%。
[0066]
实施例12 7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备:
[0067]
实施例1制备的3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮1kg,四氢呋喃10kg和醋酸0.624kg(4equiv.)溶解,搅拌下加入锌粉0.34kg(2.0equiv.),30℃反应2小时滤除不溶物后,所得滤液浓缩回收溶剂后加甲醇打浆得类白色固体0.66kg,收率80%。
[0068]
实施例13 7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备:
[0069]
实施例1制备的3,3-二溴-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮1kg,四氢呋喃13kg和醋酸0.3kg(1.9equiv.)溶解,搅拌下加入锌粉0.6kg(3.5equiv.),40℃反应2小时滤除不溶物后,所得滤液浓缩回收溶剂后加乙酸乙酯打浆得类白色固体0.62kg,收率75%。
[0070]
实施例14 7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的制备:
[0071]
实施例1制备的3,3-二溴-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮1kg,四氢呋喃13kg和醋酸0.8kg(5.1equiv.)溶解,搅拌下加入锌粉1.0kg(6equiv.),40℃反应2小时滤除不溶物后,所得滤液浓缩回收溶剂后加乙酸乙酯打浆得类白色固体0.56kg,收率68%。
[0072]
步骤(1)中卤代试剂种类、7-(4-溴苯甲酰基)吲哚:卤代试剂:酸:水:四氢呋喃的
质量比3,3-二氯-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮收率的影响
[0073][0074][0075]
步骤(2)中3,3-二卤代-7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮:醋酸:锌粉的摩尔比对7-(4-溴苯甲酰基)-1,3-二氢-2h-吲哚-2-酮的收率的影响
[0076]