1.本发明涉及有机电致发光技术领域,特别是涉及菲并咪唑类化合物及其制备方法和应用。
背景技术:
2.有机发光二极管(oled)显示器因其出色的性能而受到越来越多的关注,其制备技术越来越成熟。
3.目前oled屏幕中使用的蓝光材料主要为荧光材料,因为相比磷光材料,荧光材料的寿命会好很多。但是荧光材料的效率不高,一般荧光器件的理论eqe极限为5%。
4.为了提高蓝光效率,采用热活化延迟荧光材料(tadf)是比较好的方法,但是tadf材料一般会有很严重的效率滚降,这是三线态激子(t1)偶联导致的,当三线态激子存在时间较长且又不能及时上转换为单线态激子(s1)时就会导致三线态激子浓度变大产生偶联而导致效率滚降严重。
技术实现要素:
5.鉴于上述现有技术的不足,本发明的目的在于提供一种菲并咪唑类化合物,其发光过程是由更高能级的三线态激子(比如t2或t3)转换为单线态激子(s1),这个过程比t1转换到s1要快,其三线态激子不易堆积,从而很好地降低效率滚降且保持高效率。
6.本发明的技术方案如下:
7.一种菲并咪唑类化合物,具有如通式(1)所式的结构:
[0008][0009]
其中:
[0010]
r1选自非吸电性芳香基团;
[0011]
r2、r3分别独立地选自含氮供电子基团。
[0012]
本发明进一步涉及一种菲并咪唑类化合物的制备方法,包括以下步骤:
[0013]
所述使具有m1所示结构化合物、4-(4,6-二苯基-1,3,5-三嗪-2-基)苯胺和具有m2所示结构化合物反应,制备具有m3所示结构化合物;
[0014]
使具有m3所示结构化合物、具有m4所示结构化合物和具有m5所示结构化合物反应,制备具有通式(1)所式的结构的化合物;
[0015][0016]
r1选自非吸电性芳香基团;
[0017]
r2、r3分别独立地选自含氮供电子基团;
[0018]
x每次出现时,分别独立地选自f、cl或br。
[0019]
本发明进一步涉及一种蓝光材料,所述蓝光材料包含如上所述的菲并咪唑类化合物,或包含由上述制备方法制备而成的菲并咪唑类化合物。
[0020]
本发明进一步涉及一种发光二极管,所述发光二极管包含发光层,所述发光层包含如上所述的菲并咪唑类化合物,或包含由上述制备方法制备而成的菲并咪唑类化合物,或包含如上述的蓝光材料。
[0021]
有益效果:
[0022]
本发明所述的菲并咪唑类化合物包含1h-菲并[9,10-d]咪唑结构,此结构是产生高能级三线态激子关键,同时,还包含供电子和吸电子基团,赋予材料高效率特性。所述菲并咪唑类化合物用作为蓝光材料,兼具高效率和低效率滚降的特性,降低热活化延迟荧光材料的效率滚降效应。
附图说明
[0023]
图1为有机发光二极管元器件的结构示意图。
具体实施方式
[0024]
本发明提供一种菲并咪唑类化合物及其制备方法和应用。为使本发明的目的、技术方案及效果更加清楚、明确,以下对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
[0025]
本发明涉及一种菲并咪唑类化合物,具有如通式(1)所式的结构:
[0026][0027]
其中:
[0028]
r1选自非吸电性芳香基团;
[0029]
r2、r3分别独立地选自含氮供电子基团。
[0030]
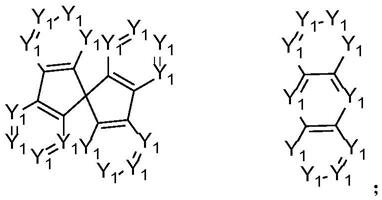
在一实施例中,所述r1选自以下基团中的一种:
[0031][0032]
其中:
[0033]
y1分别独立地选自cr4;
[0034]
r4每次出现时,分别独立选自氢、d、具有1至20个c原子的直链烷基、具有3至20个c原子的支链或环状的烷基、具有6至60个环原子的取代或未取代的芳香基、具有5至60个环原子的取代或未取代的杂芳香基。
[0035]
在一实施例中,所述r4均为氢。此时,所述r1选自以下基团中的一种:
[0036][0037]
在一实施例中,所述r2、r3分别独立地选自以下基团:
[0038][0039]
其中:
[0040]
z不存在,或选自单键、cr5r6或o;
[0041]
y2分别独立地选自cr5;
[0042]
r5、r6每次出现时,分别独立选自氢、d、具有1至20个c原子的直链烷基、具有3至20个c原子的支链或环状的烷基、具有6至60个环原子的取代或未取代的芳香基、具有5至60个环原子的取代或未取代的杂芳香基。
[0043]
在一实施例中,r5、r6每次出现时,分别独立选自氢、d、具有1至10个c原子的直链烷基、具有3至10个c原子的支链或环状的烷基。
[0044]
进一步优选地,r5、r6每次出现时,分别独立选自氢、d、具有1至5个c原子的直链烷基。
[0045]
在一实施例中,所述r2、r3分别独立地选自以下基团中的一种:
[0046][0047]
在一实施例中,所述r2和r3相同。
[0048]
在一实施例中,所述菲并咪唑类化合物选自以下结构中的一种:
[0049][0050]
其中:y1、r4、z、y2、r5和r6如上所述。
[0051]
下面列出按照本发明所述的菲并咪唑类化合物的结构,但不限于此:
[0052][0053]
[0054]
本发明的菲并咪唑类化合物,可作为蓝光材料,应用于有机电子器件,特别是oled器件中。
[0055]
本发明所述的有机电子器件可选于,但不限于,有机发光二极管(oled),有机光伏电池(opv),有机发光电池(oleec),有机场效应管(ofet),有机发光场效应管,有机激光器,有机自旋电子器件,有机传感器及有机等离激元发射二极管(organic plasmon emitting diode)等,特别优选为oled。本发明实施例中,优选将所述菲并咪唑类化合物用于oled器件的发光层。
[0056]
在一实施例中,本发明所述的有机电子器件选自溶液型有机电子器件;其部分功能层通过打印或涂布方式制备而成;在一实施例中,所述的发光层通过打印或涂布方式制备而成。
[0057]
在以上所述的发光器件,特别是oled中,包括一基片,一阳极,至少一发光层,一阴极。
[0058]
基片可以是不透明或透明。一个透明的基板可以用来制造一个透明的发光元器件。例如可参见,bulovic等nature 1996,380,p29,和gu等,appl.phys.lett.1996,68,p2606。基片可以是刚性的或弹性的。基片可以是塑料,金属,半导体晶片或玻璃。最好是基片有一个平滑的表面。无表面缺陷的基板是特别理想的选择。在一个优选的实施例中,基片是柔性的,可选于聚合物薄膜或塑料,其玻璃化温度tg为150℃以上,较好是超过200℃,更好是超过250℃,最好是超过300℃。合适的柔性基板的例子有聚(对苯二甲酸乙二醇酯)(pet)和聚乙二醇(2,6-萘)(pen)。
[0059]
阳极可包括一导电金属或金属氧化物,或导电聚合物。阳极可以容易地注入空穴到空穴注入层(hil)或空穴传输层(htl)或发光层中。在一个的实施例中,阳极的功函数和发光层中的发光体或作为hil或htl或电子阻挡层(ebl)的p型半导体材料的homo能级或价带能级的差的绝对值小于0.5ev,较好是小于0.3ev,最好是小于0.2ev。阳极材料的例子包括但不限于:al、cu、au、ag、mg、fe、co、ni、mn、pd、pt、ito、铝掺杂氧化锌(azo)等。其他合适的阳极材料是已知的,本领域普通技术人员可容易地选择使用。阳极材料可以使用任何合适的技术沉积,如一合适的物理气相沉积法,包括射频磁控溅射,真空热蒸发,电子束(e-beam)等。在某些实施例中,阳极是图案结构化的。图案化的ito导电基板可在市场上买到,并且可以用来制备根据本发明的器件。
[0060]
阴极可包括一导电金属或金属氧化物。阴极可以容易地注入电子到eil或etl或直接到发光层中。在一个的实施例中,阴极的功函数和发光层中发光体或作为电子注入层(eil)或电子传输层(etl)或空穴阻挡层(hbl)的n型半导体材料的lumo能级或导带能级的差的绝对值小于0.5ev,较好是小于0.3ev,最好是小于0.2ev。原则上,所有可用作oled的阴极的材料都可能作为本发明器件的阴极材料。阴极材料的例子包括但不限于:al、au、ag、ca、ba、mg、lif/al、mgag合金、baf2/al、cu、fe、co、ni、mn、pd、pt、ito等。阴极材料可以使用任何合适的技术沉积,如一合适的物理气相沉积法,包括射频磁控溅射,真空热蒸发,电子束(e-beam)等。
[0061]
oled还可以包含其他功能层,如空穴注入层(hil)、空穴传输层(htl)、电子阻挡层(ebl)、电子注入层(eil)、电子传输层(etl)、空穴阻挡层(hbl)。
[0062]
本发明还涉及按照本发明的有机电子器件在各种电子设备中的应用,包括,但不
限于,显示设备,照明设备,光源,传感器等等。
[0063]
本发明还涉及包含有按照本发明的有机电子器件的电子设备,包括,但不限于,显示设备,照明设备,光源,传感器等等。
[0064]
本发明所述菲并咪唑类化合物的通用合成路线如下所示:
[0065][0066][0067]
其中,x每次出现时,分别独立地选自f、cl或br。
[0068]
步骤1中间体m3的制备
[0069]
于100ml的两口瓶中依次加入2mmol m1、10mmol 4-(4,6-二苯基-1,3,5-三嗪-2-基)苯胺、2mmol m2、8mmol醋酸铵,然后加入醋酸30ml,氮气氛围下140℃回流12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取3次,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物m3。
[0070]
步骤2最终产物(1)的制备
[0071]
于150ml的两口瓶中依次加入4mmol m3、4.5mmol m4、4.5mmol m5、0.2mmol四三苯基磷钯pd(pph3)4、8mmol碳酸钾k2co3,氮气氛围下,加入混合溶剂甲苯/乙醇/纯水(v/v/v=8:1:1)80ml,然后120℃回流反应12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取3次,有机层连续用浓盐水和纯水清洗,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物(1)。
[0072]
具体实施例
[0073]
1、化合物的合成
[0074]
实施例1化合物n1及其制备方法
[0075][0076]
于100ml的两口瓶中依次加入2mmol 3,6-二溴菲醌(m1)、10mmol 4-(4,6-二苯基-1,3,5-三嗪-2-基)苯胺、2mmol 2-甲醛基-9,9'-螺二芴(m2-1)、8mmol醋酸铵,然后加入醋酸30ml,氮气氛围下140℃回流12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取3次,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物m3-1,产率71%,使用核磁氢谱来识别该化合物,1h nmr(500mhz,cd2cl2),δ(tms,ppm):9.10(s,2h),8.36(m,4h),7.96
–
8.09(m,5h),7.77
–
7.89(m,9h),7.27
–
7.50(m,15h).
[0077][0078]
于150ml的两口瓶中依次加入4mmol m3-1、9mmol 4-(9h-咔唑-9-基)苯硼酸(m4-1)、0.2mmol四三苯基磷钯pd(pph3)4、8mmol碳酸钾k2co3,氮气氛围下,加入混合溶剂甲苯/乙醇/纯水(v/v/v=8:1:1)80ml,然后120℃回流反应12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取3次,有机层连续用浓盐水和纯水清洗,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物n1,产率83%,使用核磁氢谱来识别该化合物,1h nmr(500mhz,cd2cl2),δ(tms,ppm):9.12(s,2h),8.87(d,1h),8.33
–
8.43(m,7h),7.99
–
8.01(m,7h),7.77
–
7.80(m,4h),7.50
–
7.54(m,8h).
[0079]
实施例2化合物n2其制备方法
[0080]
m3-1的制备方法与实施例1相同
[0081][0082]
于150ml的两口瓶中依次加入4mmol m3-1、9mmol m4-2、0.2mmol四三苯基磷钯pd(pph3)4、8mmol碳酸钾k2co3,氮气氛围下,加入混合溶剂甲苯/乙醇/纯水(v/v/v=8:1:1)80ml,然后120℃回流反应12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取3次,有机层连续用浓盐水和纯水清洗,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物n2,产率86%,使用核磁氢谱来识别该化合物,1h nmr(500mhz,cd2cl2),δ(tms,ppm):9.23(s,2h),8.08-8.28(m,8h),7.41-7.93(m,29h),6.69-6.92(m,20h).
[0083]
实施例3化合物n3其制备方法
[0084]
m3-1的制备方法与实施例1相同
[0085][0086]
于150ml的两口瓶中依次加入4mmol m3-1、9mmol m4-3、0.2mmol四三苯基磷钯pd(pph3)4、8mmol碳酸钾k2co3,氮气氛围下,加入混合溶剂甲苯/乙醇/纯水(v/v/v=8:1:1)80ml,然后120℃回流反应12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取3次,有机层连续用浓盐水和纯水清洗,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物n3,产率80%,使用核磁氢谱来识别该化合物,1h nmr(500mhz,cd2cl2),δ(tms,ppm):9.21(s,2h),8.15-8.27(m,6h),7.75-8.04(m,12h),7.19-7.51(m,27h),6.63-6.81(m,16h).
[0087]
实施例4化合物n4其制备方法
[0088]
m3-1的制备方法与实施例1相同
[0089][0090]
于150ml的两口瓶中依次加入4mmol m3-1、9mmol m4-4、0.2mmol四三苯基磷钯pd(pph3)4、8mmol碳酸钾k2co3,氮气氛围下,加入混合溶剂甲苯/乙醇/纯水(v/v/v=8:1:1)80ml,然后120℃回流反应12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取3次,有机层连续用浓盐水和纯水清洗,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物n4,产率59%,使用核磁氢谱来识别该化合物,1h nmr(500mhz,cd2cl2),δ(tms,ppm):9.20(s,2h),8.04-8.28(m,8h),7.75-7.93(m,10h),7.51-7.54(m,9h),7.19-7.41(m,10h),7.02-7.05(m,8h),6.55-6.73(m,12h),1.72(s,12h).
[0091]
实施例5化合物n5其制备方法
[0092][0093]
于100ml的两口瓶中依次加入2mmol 3,6-二溴菲醌(m1)、10mmol 4-(4,6-二苯基-1,3,5-三嗪-2-基)苯胺、2mmol 2-甲醛基蒽(m2-2)、8mmol醋酸铵,然后加入醋酸30ml,氮气氛围下140℃回流12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取3次,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物m3-2,产率76%,使用核磁氢谱来识别该化合物,1h nmr(500mhz,cd2cl2),δ(tms,ppm):9.23(s,2h),8.83(d,1h),8.19
–
8.55(m,15h),7.91
–
8.02(m,13h),7.72
–
7.79(m,4h),7.50
–
7.58(m,12h),7.35(m,2h),7.16-7.20(m,4h).
[0094][0095]
于150ml的两口瓶中依次加入4mmol m3-1、9mmol 4-(9h-咔唑-9-基)苯硼酸(m4-1)、0.2mmol四三苯基磷钯pd(pph3)4、8mmol碳酸钾k2co3,氮气氛围下,加入混合溶剂甲苯/乙醇/纯水(v/v/v=8:1:1)80ml,然后120℃回流反应12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取3次,有机层连续用浓盐水和纯水清洗,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物n5,产率88%,使用核磁氢谱来识别该化合物,1h nmr(500mhz,cd2cl2),δ(tms,ppm):9.27(d,2h),8.55(d,2h),8.30
–
8.37(m,8h),8.09
–
8.19(m,3h),7.89
–
7.94(m,14h),7.77
–
7.80(m,5h),7.16
–
7.50(m,25h).
[0096]
实施例6化合物n6其制备方法
[0097]
m3-2的制备方法与实施例5相同
[0098][0099]
于150ml的两口瓶中依次加入4mmol m3-2、9mmol m4-2、0.2mmol四三苯基磷钯pd(pph3)4、8mmol碳酸钾k2co3,氮气氛围下,加入混合溶剂甲苯/乙醇/纯水(v/v/v=8:1:1)80ml,然后120℃回流反应12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取3次,有机层连续用浓盐水和纯水清洗,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物n6,产率63%,使用核磁氢谱来识别该化合物,1h nmr(500mhz,cd2cl2),δ(tms,ppm):9.22(s,2h),8.13
–
8.31(m,9h),7.79
–
7.91(m,7h),7.39
–
7.54(m,15h),6.59
–
6.92(m,20h).
[0100]
实施例7化合物n7其制备方法
[0101]
m3-2的制备方法与实施例5相同
[0102][0103]
于150ml的两口瓶中依次加入4mmol m3-2、9mmol m4-3、0.2mmol四三苯基磷钯pd(pph3)4、8mmol碳酸钾k2co3,氮气氛围下,加入混合溶剂甲苯/乙醇/纯水(v/v/v=8:1:1)80ml,然后120℃回流反应12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取3次,有机层连续用浓盐水和纯水清洗,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物n7,产率78%,使用核磁氢谱来识别该化合物,1h nmr(500mhz,cd2cl2),δ(tms,ppm):9.21(s,2h),8.15
–
8.28(m,9h),7.79
–
8.04(m,7h),7.20
–
7.55(m,23h),6.63
–
6.81(m,16h).
[0104]
实施例8化合物n8其制备方法
[0105]
m3-2的制备方法与实施例5相同
[0106][0107]
于150ml的两口瓶中依次加入4mmol m3-2、9mmol m4-4、0.2mmol四三苯基磷钯pd(pph3)4、8mmol碳酸钾k2co3,氮气氛围下,加入混合溶剂甲苯/乙醇/纯水(v/v/v=8:1:1)80ml,然后120℃回流反应12h;反应完成后冷却到室温,将反应液倒入水中,用二氯甲烷萃取3次,有机层连续用浓盐水和纯水清洗,然后用无水mgso4干燥,过滤,旋蒸除去溶剂,用硅胶色谱柱进行分离提纯,用正己烷/二氯甲烷作洗脱剂,旋蒸除去溶剂收集产物n8,产率69%,使用核磁氢谱来识别该化合物,1h nmr(500mhz,cd2cl2),δ(tms,ppm):9.20(s,2h),7.91
–
8.31(m,14h),7.39
–
7.79(m,17h),7.02
–
7.05(m,8h),6.55
–
6.73(m,12h),1.69(s,12h).
[0108]
2、有机发光二极管元器件及其制备
[0109]
一种有机发光二极管元器件,其包含:第一电极、在所述第一电极上形成的空穴注入层、在所述空穴注入层上形成空穴传输层、在所述空穴传输层上形成电子阻挡层、在所述电子阻挡层上形成发光层、在所述发光层上形成的电子传输层、在所述电子传输层上形成
的电子注入层、覆盖在所述电子注入层上的第二电极;请参见图1,为ito/hil/htl/ebl/eml/etl/eil/阴极的多层有机发光二极管器件示例。结构为:ito/hat-cn(10nm)/npb(30nm)/tcta(10nm)/cbp:5wt%n1(30nm)/lg201:liq(1:1,20nm)/lif(1nm)/al(100nm)。
[0110]
其中,hat-cn作为空穴注入层(hil),npb作为空穴传输层(htl),tcta作为电子阻挡层(ebl),cbp作为主体材料,n1作为发光材料,lg201和liq共同作为电子传输层(etl),lif作为电子注入层(eil),al作为阴极。该示例器件记作“n1器件”。
[0111]
该有机发光二极管元器件的制备方法包括如下步骤:
[0112]
(1)首先对ito基板按如下次序进行清洗:5%koh溶液超声15min、纯水超声15min、异丙醇超声15min、烘箱干燥1h;
[0113]
(2)然后将基板转移至uv-ozone设备进行表面处理15min,处理完后立即转移至手套箱中。
[0114]
(3)随后进行蒸镀成膜:依次制备空穴注入层、空穴传输层、电子阻挡层、发光层、电子传输层、电子注入层、第二电极;先抽真空至10-7
torr,然后缓慢提高电流值,使速率缓慢增加至待速率稳定后打开挡板进行蒸镀。
[0115]
(4)最后进行uv固化封装,再80℃烘烤60min即可。
[0116]
参照该实施例的方法,采用化合物n2~n8作为发光材料制备图1示例的器件,分别记作“n2器件”、“n3器件”,
……
,“n8器件”。
[0117]
参照该实施例的方法,采用常见的tadf材料dcz-trz:制备图1示例的器件,记作“r1器件”。结构为:ito/hat-cn(10nm)/npb(30nm)/tcta(10nm)/cbp:5wt%dcz-trz(30nm)/lg201:liq(1:1,20nm)/lif(1nm)/al(100nm)。
[0118]
参照常规方法,对n1器件至n8器件及r1器件进行测试,获得结果参见表1。
[0119]
表1
[0120]
器件最大外量子效率[%]
*
效率滚降r17.7863.8%n19.326.5%n28.4214.6%n37.8712.2%n48.3318.9%n59.0212.3%n68.4524.6%
n78.9516.5%n88.1126.9%
[0121]
效率滚降:最大外量子效率减去1000nits对应的外量子效率之差与最大外量子效率的比值。
[0122]
综上所述,本发明的菲并咪唑类化合物作为蓝光材料制备的发光二极管,其效率高于常见的tadf材料dcz-trz,且效率滚降明显降低。
[0123]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0124]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。