三氮唑-4-(n-取代甲酰胺)-5-三氮烯类化合物及其制备方法和应用
技术领域
1.本发明属于药物化学领域,具体涉及一种1-取代-1h-1,2,3-三氮唑-4-(n-取代甲酰胺)-5-三氮烯类化合物的制备方法,并对钠离子通道na
v 1.1的半数激活电压以及半数失活电压进行了测试。
背景技术:
2.三氮唑类化合物是具有生物活性或药用价值的一类物质。上市药物卢非酰胺具有三氮唑母核结构,用于癫痫lennox-gastaut综合征(lgs)的辅助治疗。特别地,该类化合物与卢非酰胺结构类似,具有明显电生理活性,降低钠离子通道nav1.1半数失活电压,从而缩短钠离子通道nav1.1开放时间。部分化合物在动物癫痫模型上,如戊四唑急性癫痫发作模型(ptz)、最大电休克癫痫发作模型(mes),表现出较卢非酰胺更好的抗癫痫作用。
3.
技术实现要素:
4.本发明提供了1-取代-1h-1,2,3-三氮唑-4-(n-取代甲酰胺)-5-三氮烯类化合物制备方法以及钠离子通道nav1.1生物活性评价。同时,部分化合物在动物癫痫模型以及细胞水平上钠离子通道nav1.1的半数激活电压和半数失活电压较卢非酰胺具有更好的生物活性。
5.1-取代-1h-1,2,3-三氮唑-4-(n-取代甲酰胺)-5-三氮烯类化合物,结构如式(ⅰ)所示:
[0006][0007]
式(ⅰ)中,r1为取代或者未取代的苯基、取代或者未取代的苄基、取代或者未取代的c2~c4烷基,所述苯基上的取代基为卤素、c1~c6烷基,所述苄基上的取代基为卤素、c1~c6烷基,所述c2~c4烷基上的取代基为一个或者多个氟取代的苯基;
[0008]
r2为氢、取代或者未取代的c1~c8烷基、苯基、苄基,所述c1~c8烷基上的取代基为甲氧酰基;
[0009]
r3和r4独立地选自为c1~c4烷基,或者r3、r4与连接r3和r4的n形成五元或者六元环,
所述五元环或者六元环中含有n、c和o,并且可被酰基取代。
[0010]
细胞以及动物实验表明该类结构具有比卢非酰胺更好的生物活性。
[0011]
作为优选,所述的r1为2,6-二氟苄基、2,6-二氟苯基、2-(2,6-二氟苯基)乙基、2,4-二氟苄基、2-氟苄基、4-氟苄基、苯基、苄基、2-氯苄基、4-甲基苄基、正丁基;r2为氢、甲基、环丙基、苯基、苄基;r3和r4独立地选自甲基、异丙基或者r3、r4与连接r3和r4的n形成吡咯烷、哌啶、吗啡啉、n-甲酰基哌嗪。
[0012]
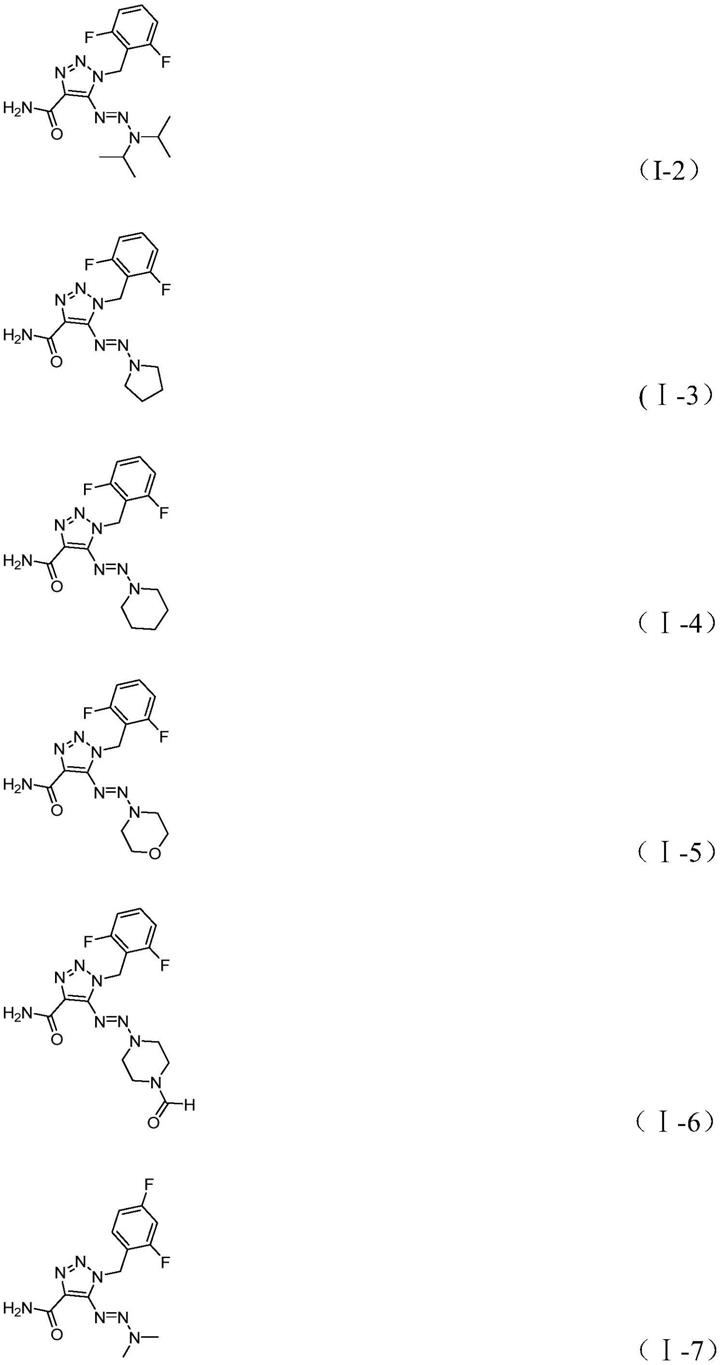
作为进一步的优选,所述的1-取代-1h-1,2,3-三氮唑-4-(n-取代甲酰胺)-5-三氮烯类化合物选自如下具体化合物中的一种:
[0013]
[0014]
[0015]
[0016][0017]
本发明还提供了一种所述的1-取代-1h-1,2,3-三氮唑-4-(n-取代甲酰胺)-5-三氮烯类化合物的制备方法,其特征在于,包括如下步骤:
[0018]
步骤1:二级胺于-78℃条件下与正丁基锂生成氨基锂,将新制备的氨基锂thf溶液通入到一氧化二氮气体氛围中反应4h;
[0019]
步骤2:在上述反应期间,将2-(2-炔丙氧基)四氢吡喃与乙基溴化镁试剂反应2h。将现制备1-(2-四氢吡喃氧基)丙炔溴化镁加入到步骤1反应中50℃反应过夜,得到中间体1;
[0020]
步骤3:中间体1在1mol%[ir(cod)cl]2催化剂下与烷基叠氮生成中间体2;
[0021]
步骤4:中间体2在meoh/hcl(ph=2~3)酸性条件下生成中间体3。中间体3溶于pbs(ph=6.8)/乙腈(v/v=1:1)中,在3eq naclo2/10mol%tempo/40℃条件下生成中间体4;
[0022]
步骤5:中间体4与当量cdi室温反应30min,随后与有机胺室温反应生成1-取代-1h-1,2,3-三氮唑-4-(n-取代甲酰胺)-5-三氮烯类化合物。
[0023]
所述的中间体1的结构如式(ⅱ)所示:
[0024][0025]
所述的中间体2结构如式(ⅲ)所示:
[0026][0027]
所述的中间体3结构如式(ⅳ)所示
[0028][0029]
所述的中间体4结构如式(
ⅴ
)所示
[0030][0031]
其中,r1、r2、r3和r4的定义如前文所述。
[0032]
反应过程中,二级胺与正丁基锂反应生成氨基锂,后者在n2o氛围中反应一段时间,之后和1-(2-四氢吡喃氧基)丙炔溴化镁反应生成三氮烯衍生物中间体1。三氮烯中间体1在[ir(cod)cl]2催化剂作用下与烷基叠氮生成三氮唑衍生物中间体2。三氮唑衍生物中间体2在酸性条件下脱除thp保护基得到中间体3,随后在naclo2/10mol%tempo体系下将中间体中羟基氧化成酸生成中间体4。最后,中间体4羧基被cdi活化,之后与胺缩合反应生成目标化合物。
[0033]
作为优选,所述的铱催化剂为[ir(cod)cl]2(cas:12112-67-3)用量为所述的三氮烯类化合物的摩尔量的1~2%,该种铱催化剂对本发明中的底物的适用性好,催化效率高。
[0034]
作为优选,所述的1-(2-四氢吡喃氧基)丙炔溴化镁,通过四氢-2-(2-丙炔氧基)-2h-吡喃与乙基溴化镁反应得到,用量为二级胺底物的1.5eq。
[0035]
作为优选,所述的中间体3中的羟基氧化成中间体4,最优条件:naclo2为3eq,10mol%tempo催化量,反应溶剂为pbs/乙腈1:1混合溶剂,其中pbs缓冲液ph=6.8,40℃。
[0036]
作为优选,所述的r1为2,6-二氟苄基、2,6-二氟苯基、2-(2,6-二氟苯基)乙基、2,4-二氟苄基、2-氟苄基、4-氟苄基、苯基、苄基、2-氯苄基、4-甲基苄基、正丁基。
[0037]
作为优选,所述的r2为氢、甲基、环丙基、苯基、苄基。
[0038]
作为优选,所述的r3和r4独立地选自甲基、异丙基或者r3、r4与连接r3和r4的n形成吡咯烷、哌啶、吗啡啉、n-甲酰基哌嗪.
[0039]
作为优选,所述的三氮烯中间体1类化合物可以参照现有方法制备,具体可参见文献(angew.chem.int.ed.2015,54,302
–
305)。
[0040]
作为优选,所述的化合物能够明显降低钠离子通道nav1.1半数失活电压,从而缩短钠离子通道nav1.1开放时间。
[0041]
作为优选,所述的化合物在动物模型上,如戊四唑急性癫痫发作模型(ptz)、最大电休克癫痫发作模型(mes),部分化合物表现出较卢非酰胺更好的抗癫痫作用。
附图说明
[0042]
图1为实施例1制得的产物的1hnmr谱图;
[0043]
图2为实施例1制得的产物的
13
cnmr谱图;
[0044]
图3为实施例2制得的产物的1hnmr谱图;
[0045]
图4为实施例2制得的产物的
13
cnmr谱图;
[0046]
图5为实施例23测得的化合物z-1给药前后的nav1.1钠通道电流激活和失活曲线;
[0047]
图6为实施例23测得的化合物z-2给药前后的nav1.1钠通道电流激活和失活曲线;
[0048]
图7为实施例23测得的化合物z-3给药前后的nav1.1钠通道电流激活和失活曲线;
[0049]
图8为实施例23测得的化合物z-4给药前后的nav1.1钠通道电流激活和失活曲线;
[0050]
图9为实施例23测得的化合物z-5给药前后的nav1.1钠通道电流激活和失活曲线;
[0051]
图10为实施例23测得的化合物z-6给药前后的nav1.1钠通道电流激活和失活曲线;
[0052]
图11为实施例23测得的化合物z-7给药前后的nav1.1钠通道电流激活和失活曲线;
[0053]
图12为实施例23测得的化合物z-8给药前后的nav1.1钠通道电流激活和失活曲线;
[0054]
图13为实施例23测得的化合物z-9给药前后的nav1.1钠通道电流激活和失活曲线;
[0055]
图14为实施例23测得的化合物z-10给药前后的nav1.1钠通道电流激活和失活曲线;
[0056]
图15为实施例23测得的化合物z-10给药前后的nav1.1钠通道电流激活和失活曲线;
[0057]
图16为实施例23测得的化合物z-12给药前后的nav1.1钠通道电流激活和失活曲线;
[0058]
图17为实施例23测得的化合物z-13给药前后的nav1.1钠通道电流激活和失活曲线;
[0059]
图18为实施例23测得的化合物z-14给药前后的nav1.1钠通道电流激活和失活曲线;
[0060]
图19为实施例23测得的化合物z-15给药前后的nav1.1钠通道电流激活和失活曲线;
[0061]
图20为实施例23测得的化合物z-16给药前后的nav1.1钠通道电流激活和失活曲线;
[0062]
图21为实施例24中部分化合物对戊四唑急性癫痫发作模型(ptz)的作用:
[0063]
图22为实施例24中部分化合物对最大电休克癫痫发作模型(mes)的作用。
具体实施方式
[0064]
下面结合具体实施例对本发明做详细的描述,这些实施例只是用于解释本发明的技术方案,并不是对本发明形成任何的限制。
[0065]
本发明中所用的1-取代-1h-1,2,3-三氮唑-4-(n-取代甲酰胺)-5-三氮烯类化合物可以采用如下方法进行制备:
[0066][0067]
实施例1
[0068][0069]
步骤1:将15mmol二异丙胺加入到两口瓶中,氩气保护,加入15ml无水thf。于-78℃条件下,缓慢滴加6.6ml正丁基(2.5m,16.5mmol),滴加结束后继续反应30min,随后移至室温。将新制备的二异丙基氨基锂thf溶液通入到n2o气体氛围中,补加15ml无水thf,rt反应4h;
[0070]
步骤2:在上述反应期间,将2.8g 2-(2-炔丙氧基)四氢吡喃(20mmol)加入到二口瓶中,氩气保护,加入20ml无水thf。于室温条件下,缓慢滴加20ml ch3mgbr试剂(1m),滴加结束后50℃反应2h;
[0071]
步骤3:步骤1反应结束后,将n2o气体置换成氩气保护。随后将步骤2反应液通过注射器转移到步骤1反应体系中,50℃反应过夜。反应结束后,加30ml水淬灭,硅藻土抽滤,乙酸乙酯萃取三次,饱和nacl水洗一次,无水na2so4干燥,减压除去溶剂。使用中性al2o3为固定相、石油醚/乙酸乙酯体系为洗脱剂进行柱色谱分离纯化,得到2.5g白色固体中间体1,收率62%;
[0072]
步骤4:在25ml单口反应瓶中,,加入1g中间体1(3.7mmol)、633mg2-(叠氮甲基)-1,3-二氟苯(3.7mmol)、25mg[ir(cod)cl]2催化剂(1mol%,0.037mmol)。加入10ml二氯甲烷,
室温反应。反应结束后,使用硅胶为固定相,石油醚/乙酸乙酯体系为洗脱剂进行柱色谱分离纯化。得到1.1g白色固体中间体2,收率68%。
[0073]
步骤5:在50ml单口瓶中,加入1.1g中间体2,加入10ml meoh,用盐酸将反应液调至ph=2~3,室温反应。反应结束后,将反应液中甲醇减压旋出,得到中间体3。将中间体3溶于10ml pbs(ph=6.8)/10ml乙腈(v/v=1:1)中,室温下加入680mg naclo2、40mg tempo、1~2滴6-14%naclo溶液,40℃反应。反应结束后,用naso3溶液淬灭反应液中剩余的naclo2,随后用1n naoh溶液将反应液调至碱性。将反应液中乙腈减压旋出,乙醚萃取三次,弃有机层保留水层。将水层用盐酸调至酸性,析出大量白色固体,过滤,烘干,得到560mg白色固体中间体4,两步收率61%;
[0074]
步骤6:在25ml单口瓶中,加入560mg(1.5mmol)中间体4、290mg cdi(1.8mmol),加入5ml乙腈,室温反应30min。tlc检测中间体4反应完全后,加入0.5ml 28-30%氨水,继续室温反应。反应结束后,将反应液中乙腈减压旋出。粗产物溶于15ml二氯甲烷中,1m hcl水洗三次,饱和nacl水洗一次,无水na2so4干燥,减压除去溶剂。二氯甲烷/甲醇洗脱剂进行柱色谱分离纯化得到目标产物,白色固体420mg,收率76%。
[0075]
产品物理性质及谱图数据如下:白色固体;mp:166.0-166.2℃;1h nmr(400mhz,cdcl3)δ7.34-7.26(m,1h),6.92-6.86(m,2h),5.64(s,2h),5.32-5.26(m,1h),4.20-4.10(m,1h),1.39(d,j=6.8hz,3h),1.29(d,j=6.8hz,3h);
13
c nmr(101mhz,cdcl3)δ161.48(dd,j=251.6,7.2hz),161.15(s),146.79(s),130.86(t,j=10.3hz),125.53(s),111.73-111.49(m),110.87(t,j=18.4hz),51.01(s),48.49(s),39.77(s),23.09(s),18.79(s);hrms(ei)(m/z):calcd for 366.1854(m
+
),366.1854;found,366.1851。
[0076]
产物的1hnmr谱图见图1,
13
cnmr谱图见图2。
[0077]
实施例2
[0078][0079]
步骤1:将20mmol二甲胺加入到两口瓶中,氩气保护,加入15ml无水thf。于-78℃条件下,缓慢滴加8.8ml正丁基(2.5m,22mmol),滴加结束后继续反应30min,随后移至室温。将新制备的二甲氨基锂thf溶液通入到n2o气体氛围中,补加20ml无水thf,rt反应4h;
[0080]
步骤2:在上述反应期间,将4.2g 2-(2-炔丙氧基)四氢吡喃(30mmol)加入到二口瓶中,氩气保护,加入30ml无水thf。于室温条件下,缓慢滴加30ml ch3mgbr试剂(1m),滴加结束后50℃反应2h;’[0081]
步骤3:步骤1反应结束后,将n2o气体置换成氩气保护。随后将步骤2反应液通过注射器转移到步骤1反应体系中,50℃反应过夜。反应结束后,加50ml水淬灭,硅藻土抽滤,乙酸乙酯萃取三次,饱和nacl水洗一次,无水na2so4干燥,减压除去溶剂。使用中性al2o3为固定相、石油醚/乙酸乙酯体系为洗脱剂进行柱色谱分离纯化,得到780mg油状液体中间体1,收率18.5%;
[0082]
步骤4:在25ml单口反应瓶中,,加入300mg中间体1(1.4mmol)、260mg 2-(叠氮乙基)-1,3-二氟苯(1.4mmol)、9.5mg[ir(cod)cl]2催化剂(1mol%,0.014mmol)。加入5ml二氯甲烷,室温反应。反应结束后,使用硅胶为固定相,石油醚/乙酸乙酯体系为洗脱剂进行柱色谱分离纯化。得到450mg红色稠状固体中间体2,收率82%。
[0083]
步骤5:在25ml单口瓶中,加入450mg中间体2,加入5ml meoh,用盐酸将反应液调至ph=2~3,室温反应。反应结束后,将反应液中甲醇减压旋出,得到中间体3。将中间体3溶于5ml pbs(ph=6.8)/5ml乙腈(v/v=1:1)中,室温下加入311mg naclo2、18mg tempo、1~2滴6-14%naclo溶液,40℃反应。反应结束后,用naso3溶液淬灭反应液中剩余的naclo2,随后用1n naoh溶液将反应液调至碱性。将反应液中乙腈减压旋出,乙醚萃取三次,弃有机层保留水层。将水层用盐酸调至酸性,析出大量白色固体,过滤,烘干,得到210mg白色固体中间体4,两步收率56%;
[0084]
步骤6:在25ml单口瓶中,加入97mg(0.3mmol)中间体4、54mg cdi(0.33mmol),加入3ml乙腈,室温反应30min。tlc检测中间体4反应完全后,加入0.25ml 28-30%氨水,继续室温反应。反应结束后,将反应液中乙腈减压旋出。粗产物溶于15ml二氯甲烷中,1m hcl水洗三次,饱和nacl水洗一次,无水na2so4干燥,减压除去溶剂。二氯甲烷/甲醇洗脱剂进行柱色谱分离纯化得到目标产物,白色固体80mg,收率83%。
[0085]
产品物理性质及谱图数据如下:白色固体;mp:191.5-192.0℃;1h nmr(500mhz,cdcl3)δ7.21-7.15(m,2h),7.06-6.77(m,2h)5.52(s,1h),4.88-4.38(m,1h),3.60(d,j=4.7hz,3h),3.38-3.17(m,5h);
13
c nmr(126mhz,cdcl3)δ162.75,161.80(dd,j=247.9,8.2hz),145.33,129.58,128.93-128.71(m),113.09(t,j=18.8hz),111.30(dd,j=20.2,5.8hz),47.14,44.02,36.61,23.20;hrms(ei)(m/z):calcd for c
13h16
f2n7o(m
+
),324.1384;found,324.1378。
[0086]
产物的1hnmr谱图见图3,
13
cnmr谱图见图4。
[0087]
实施例3~22
[0088][0089]
实施例3~22的制备方法与实施例1和例2相同,不同之处在于将r1、r2、r3片段底物替换成其他相似结构底物原料,得到的产物结构列于表1。
[0090]
表1实施例3~22的产物结构
[0091]
[0092]
[0093]
[0094][0095]
实施例23 1-取代-1h-1,2,3-三氮唑-4-(n-取代甲酰胺)-5-三氮烯类化合物钠离子通道nav1.1生物活性
[0096]
2.1测试化合物信息
[0097]
表2测试化合物信息
[0098]
[0099]
[0100][0101]
2.2溶媒
[0102]
名称:dmso(二甲亚砜)
[0103]
来源:sigma购买,货号:276855-500ml
[0104]
分子量:78.13
[0105]
保存条件:常温密封避光保存
[0106]
2.3细胞信息
[0107]
种属&品系:nav1.1-flp-in
tm t-rex
tm
293细胞系
[0108]
(稳定表达nav1.1通道的flp-in
tm t-rex
tm
293细胞)
[0109]
来源:内部构建
[0110]
培养条件:5%co2,37℃培养箱
[0111]
冻存条件:液氮
[0112]
2.4溶液及试剂信息
[0113]
细胞外液配方(mm):140nacl、5kcl、1cacl2、1.25mgcl2、10hepes、10glucose,用naoh调节ph至7.4。
[0114]
细胞内液配方(mm):130csf、10nacl、10egta、10hepes,用csoh调节ph至7.2。
[0115]
缩写注释
[0116]
hepes:4-(2-羟乙基)哌嗪-1-乙磺酸,n-(2-羟乙基)哌嗪-n
′‑
(2-乙磺酸)
[0117]
egta:乙二醇双(2-氨基乙基醚)四乙酸
[0118][0119]
[0120]
2.5实验仪器
[0121]
膜片钳放大器(axopatch 200b,axon,美国)
[0122]
数模转换器(digidata 1550b,axon,美国)
[0123]
倒置显微镜(ix51,olympus,日本)
[0124]
快速给药系统(rsc-200,bio-logic,法国)
[0125]
微操作器(mx7600r,syskiyou,美国)
[0126]
电极拉制仪(p-97,sutter,美国)
[0127]
玻璃电极(bf150-86-10,sutter,美国)
[0128]
防震台与屏蔽网(63-534,tmc,美国)
[0129]
数据采集与分析软件(pclamp 11,axon,美国)
[0130]
二氧化碳培养箱(heracell 150i,thermo,美国)
[0131]
生物安全柜(model 1384,thermo,美国)
[0132]
纯水仪(milli q,millipore,美国)
[0133]
2.6实验方法
[0134]
1.化合物准备
[0135]
化合物用dmso配制成100mm的母液,储存于-20℃。试验当天,将化合物dmso母液在室温解冻后,用细胞外液稀释成所需测试的最终检测浓度。
[0136]
2.细胞培养和处理
[0137]
稳定表达nav1.1的flp-in
tm
t-rex
tm
293细胞培养于直径35mm的细胞培养皿中,置于37℃,5%co2的培养箱培养,每48小时按1:4比例进行传代,培养基配方:90%dmem(invitrogen),10%胎牛血清(gibco)和50g/ml hygromycin b(invitrogen)。试验当天,吸走细胞培养液,用细胞外液淋洗一遍后加入0.25%trypsin-edta(invitrogen)溶液,在室温下消化1分钟。吸走消化液,用细胞外液重悬后将细胞转移到用于电生理记录的实验皿中备用。
[0138]
3.电生理记录过程
[0139]
稳定表达nav1.1钠通道的flp-in
tm
t-rex
tm
293细胞,在室温下用全细胞膜片钳技术记录nav1.1钠通道电流。玻璃微电极由玻璃电极毛胚(bf150-86-10,sutter)经拉制仪拉制而成,灌注电极内液后的尖端电阻为2-5mω左右,将玻璃微电极插入放大器探头即可连接至膜片钳放大器。钳制电压和数据记录由pclamp 11软件通过电脑控制和记录,采样频率为20khz,滤波频率为2khz。在得到全细胞记录后,细胞钳制在-120mv,通过给予20ms的不同去极化电压刺激(从-120mv至+60mv,每个电压步阶间隔10mv)记录通道激活过程并计算半数激活电压(v1/2,即通道激活达到最大电导50%时的电压)。nav1.1钠通道失活过程通过给予从-120mv到+50mv(每个电压步阶间隔10mv,持续200ms)的不同条件电压,然后再给予一个-20mv,20ms的电压刺激诱发不同条件电压下的钠通道电流以记录通道失活过程并计算半数失活电压(v
1/2
,即通道失活达到最大电导50%时的电压)。化合物给药1分钟后再次记录通道激活和失活过程并计算v
1/2
,至少测试3个细胞(n≥3)。
[0140]
4.数据处理
[0141]
数据分析处理采用pclamp 11,graphpad prism 5和excel软件。电压依赖的激活曲线和失活曲线由boltzmann方程拟合计算v
1/2
:
[0142]
y=bottom+(top-bottom)/(1+exp((v
1/2-x)/slope))
[0143]
其中,x代表去极化电压,y代表去极化电压下相应的电导,bottom和top分别为最小和最大电导值。
[0144]
5.质量控制
[0145]
报告中的实验数据满足以下质量控制指标:
[0146]
全细胞封接阻抗》1gω
[0147]
串联电阻如大于10mω补偿80%以上
[0148]
给药前电流无明显的自发性衰减或增大
[0149]
6.测试结果
[0150]
化合物给药前后nav1.1通道电流激活(activation)和失活(inactivation)的δv
1/2
和δv
1/2
。
[0151]
表3化合物给药前后nav1.1通道δv
1/2
和δv
1/2
[0152][0153]
测试化合物给药前后的nav1.1钠通道电流激活和失活曲线
[0154]
[0155]
[0156][0157]
实施例24 1-取代-1h-1,2,3-三氮唑-4-(n-取代甲酰胺)-5-三氮烯类化合物动物实验
[0158]
3.1戊四唑急性癫痫发作模型(ptz)
[0159]
优选化合物z-2、z-3、z-6对戊四唑急性癫痫发作模型(ptz)的作用:采用成年雄性icr小鼠,体重25-30g。将小鼠按照体重随机分组:溶剂组、rufinamide阳性对照组以及不同化合物(剂量均为50mg/kg)组,每组5-6例icr雄鼠。实验时分别灌胃给以溶剂或药物,1h后腹腔注射100mg/kg ptz,之后立即开始观察并记录小鼠的癫痫发作情况,观察时间为30分钟。行为学评价分级标准为:0级:无明显反应;1级:失神呆滞,耳朵和面部抽动;2级:全身抽动,无前肢抬起;3级:一侧或双侧前肢抬起;4级:一侧身体侧倒伴抽搐;5级:背部着地倒地,或全身强直阵挛发作;6级:强直后死亡。记录各小鼠癫痫发作的等级(图a)、从给药开始到2级发作潜时(图b)、4级发作潜时(图c)以及6级发作潜时(图d)。
[0160]
3.2最大电休克癫痫发作模型(mes)
[0161]
优选化合物z-2、z-3、z-6对最大电休克癫痫发作模型的作用:采用成年雄性icr小鼠,体重25-30g。将小鼠按照体重随机分组:溶剂组、rufinamide阳性对照组以及不同化合物(剂量均为50mg/kg)组,每组5-6例icr雄鼠。实验时分别灌胃给以溶剂或药物,1h后将最大电休克仪的刺激夹以生理盐水浸湿后,分别夹住小鼠两耳耳廓,给以电刺激,刺激频率为50hz,刺激持续时间为0.2s,刺激电流强度为25ma。小鼠行为学评定等级:1级:跑动;2级:前肢强直;3级:后肢强直。记录小鼠在此模型中最大发作等级(图a)、到达三级发作潜时(图b)
以及强直强直阵挛发作持续时间(图c)。