1.本发明涉及放射化学合成领域,尤其涉及一种放射性同位素碳-14标记的呋虫胺和中间体及其制备方法和应用。
背景技术:
2.呋虫胺,英文通用名dinotefuran,化学名称为(rs)-1-甲基-2-硝基-3-[(3-四氢呋喃)甲基]胍,是日本三井公司开发的一种新烟碱类杀虫剂,具有高效、广谱、对鸟类和哺乳动物安全等优良特性,且具有良好的内吸渗透性,用于水稻、蔬菜、果树等防治鳞翅目、半翅目、直翅目、膜翅目等害虫,开发前景广阔。
[0003]
放射性标记,是用放射性核素取代化合物分子的一种或几种原子而使它能被识别并可用作示踪剂的化合物。它与未标记的相应化合物具有相同的化学及生物学性质,不同的只是它带有放射性,因而可利用放射性探测技术来追踪。
[0004]
现有技术大多只研究了非放射性标记的呋虫胺的合成,而本发明为进一步深入研究呋虫胺的代谢过程,合成一种放射性标记的呋虫胺。
技术实现要素:
[0005]
针对现有技术存在的问题,本发明提供一种放射性同位素碳-14标记的呋虫胺和中间体及其制备方法和应用。
[0006]
第一方面,本发明提供一种放射性同位素碳-14标记的化合物im-3,其结构式如下:
[0007][0008]
其中,*表示放射性同位素碳-14标记位置。
[0009]
本发明还提供上述放射性同位素碳-14标记的化合物im-3的合成方法。
[0010]
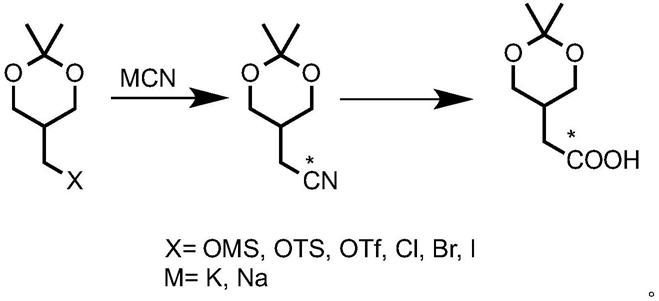
第一种方法:以2,2-二甲基-5-(卤代甲基)-1,3-二氧六环烷或1-(2,2-二甲基-1,3-二氧六环烷-5-)-甲基磺酸酯为原料,通过与碳-14标记的氰化物反应,得到2-(2,2-二甲基-1,3-二氧六环-5-)-乙腈,再经水解反应得到化合物im-3,其合成路线如下:
[0011][0012]
第二种方法:以2,2-二甲基-5-(卤代甲基)-1,3-二氧六环烷为原料,通过与镁反应得到格式试剂,再经与碳-14标记的二氧化碳反应得到化合物im-3,其合成路线如下:
[0013][0014]
第二方面,本发明提供一种放射性同位素碳-14标记的3-氨甲基四氢呋喃,其结构式如下:
[0015][0016]
其中,*表示放射性同位素碳-14标记位置。
[0017]
3-氨甲基四氢呋喃是合成呋虫胺的关键中间体,本发明合成放射性同位素碳-14标记的3-氨甲基四氢呋喃,为合成放射性同位素碳-14标记的呋虫胺提供条件。
[0018]
本发明还提供上述放射性同位素碳-14标记的3-氨甲基四氢呋喃的制备方法。
[0019]
第一种方法:以化合物im-3为原料,经过水解,关环,得到3-羟甲基丁内脂,然后制备成磺酸酯,与二苄胺发生取代反应,再经过还原,关环,脱除苄基后,得到放射性同位素碳-14标记的3-氨甲基四氢呋喃。
[0020]
第二种方法:以化合物im-3为原料,经过还原,水解后,关环制备3-羟甲基四氢呋喃,然后制备成磺酸酯,与邻苯二甲酰亚胺盐发生取代反应,脱除保护基团,得到放射性同位素碳-14标记的3-氨甲基四氢呋喃。
[0021]
第三种方法:以化合物im-3为原料,经过还原,水解后,关环制备3-羟甲基四氢呋喃,然后制备成磺酸酯,与叠氮化物进行取代反应,经过还原后,得到放射性同位素碳-14标记的3-氨甲基四氢呋喃。
[0022]
第三方面,本发明提供一种放射性同位素碳-14标记的呋虫胺,其结构式如下:
[0023][0024]
其中,*表示放射性同位素碳-14标记位置。
[0025]
本发明还提供上述放射性同位素碳-14标记的呋虫胺的制备方法,是以上述放射性同位素碳-14标记的3-氨甲基四氢呋喃为原料之一进行制备。
[0026]
具体地,可采用的方法一:以所述放射性同位素碳-14标记的3-氨甲基四氢呋喃和o-甲基-n-硝基-n
’‑
甲基异脲为原料进行制备。
[0027]
可采用的方法二:以所述放射性同位素碳-14标记的3-氨甲基四氢呋喃和s-甲基-n-硝基-n
’‑
甲基异硫脲为原料进行制备;
[0028]
可采用的方法三:以所述放射性同位素碳-14标记的3-氨甲基四氢呋喃和s-甲基-n-硝基-n
’‑
邻苯二甲酰异硫脲为原料进行制备。
[0029]
本发明提供了一种放射性同位素碳-14标记的呋虫胺和中间体及其制备方法和应用,本发明以简单的方法合成得到了化合物im-3,然后以其为原料可经多条路线合成得到关键中间体放射性同位素碳-14标记的3-氨甲基四氢呋喃,从而顺利制备得到放射性同位素碳-14标记的呋虫胺,基于同位素示踪,有助于呋虫胺的代谢研究。
附图说明
[0030]
图1为本发明实施例中所得放射性同位素碳-14标记的呋虫胺的质谱图。
具体实施方式
[0031]
为使本发明的目的、技术方案和优点更加清楚,下面对本发明中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0032]
若无特别说明,以下实施例中所涉及的原材料均可通过市购获得。
[0033]
实施例1
[0034]
本实施例提供一种放射性同位素碳-14标记的呋虫胺,其合成路线如下:
[0035][0036]
具体制备步骤如下:
[0037]
step 1
[0038]
取2.5g化合物sm-1(17.1mmol)溶于40ml dcm中,向体系中加入咪唑1.7g和碘5.3g,冷却至-5~5℃。向其中加入三苯基磷固体5.5g,室温反应3h左右。tlc检测反应完成后,加二氯甲烷和水稀释,分出有机相,无水硫酸钠干燥,浓缩,硅胶柱制备分离,得im-1纯品4.5g。
[0039]
step 2
[0040]
取3.84g im-1(15mmol)溶于20ml dmf中,向体系中加入670mgkcn(10mmol),氮气保护下于80℃反应约5h。反应完成后,确保体系为碱性,乙酸乙酯和水稀释,分出有机相,乙酸乙酯萃取。合并有机相,饱和食盐水洗涤,无水硫酸钠干燥,浓缩,硅胶柱分离,得im-2纯品1.40g。
[0041]
step 3
[0042]
取1.4g im-2(8.40mmol)溶于10ml正丁醇,向体系中加入3ml 25%naoh。氮气保护下,加热至110℃,回流12h,液相检测反应完成后,冷却至室温,用0.5m等量的稀盐酸中和,ea萃取,合并有机相,无水硫酸钠干燥,过滤,减压浓缩至干,得im-3粗品1.3g。
[0043]
step 4
[0044]
将上述制备的im-3溶于甲醇中,用3m稀盐酸调节ph值至1左右,室温搅拌2h,ea萃取,合并有机相,无水硫酸钠干燥,过滤,减压浓缩至干,得im-4粗品0.8g。
[0045]
step 5
[0046]
将0.8g im-4(6.9mmol)和对甲基苯磺酰氯(1.9g,10mmol)置于干燥的反应瓶中,氮气保护下,加入20ml二氯甲烷,搅拌溶解,冷却至-5~5℃。向反应液中滴加三乙胺的二氯甲烷溶液(1.1g,10mmol),搅拌过夜。反应完成后,反应液分别用水和饱和氯化钠溶液洗,无水硫酸钠干燥,过滤,浓缩得im-5粗品1.8g,收率95%。
[0047]
step 6
[0048]
将1.8g im-5(6.6mmol)和2g无水碳酸钾加入到反应瓶中,氮气保护下,加入20ml乙腈和1.32g二苄胺,加热回流24h,过滤浓缩,得粗品,硅胶柱层析分离,得im-6产品1.36g,收率70%。
[0049]1h nmr(400mhz,cdcl3):δ1.89-1.98(m,1h),2.31-2.39(m,1h),2.58-2.63(s,1h),2.71-2.79(m,1h),2.83-2.87(m,1h),3.38(d,2h),3.76(d,2h),4.08-4.18(m,2h),7.25-7.27(m,2h),7.30-7.36(m,8h)。
[0050]
step 7
[0051]
在氮气保护下,将380mg氢化锂铝分散于10ml无水四氢呋喃中,冷却至-5~5℃。将1.36gim-6(4.6mmol)溶于10ml无水四氢呋喃中,滴加到上述溶液中。室温反应2h,原料反应完全后,加水淬灭,过滤,滤液硫酸钠干燥,浓缩,硅胶柱分离,得im-7纯品1.12g,收率80%。
[0052]1h nmr(400mhz,cdcl3):δ1.31-1.36(m,2h),2.10-2.14(m,1h),2.37-2.41(m,1h),2.45-2.51(m,1h),3.30(d,2h),3.35-3.38(m,1h),3.52-3.58(m,3h),3.77(d,2h),7.22-7.33(m,10h)。
[0053]
step 8
[0054]
在氮气保护下,将1.12g im-7和对甲基苯磺酰氯(1.48g)溶于20ml干燥的吡啶中。室温反应48h,原料反应完全后,加水淬灭,乙醚萃取,硫酸钠干燥,过滤,浓缩,硅胶柱分离,
得im-8纯品0.74g,收率70%。
[0055]1h nmr(400mhz,cdcl3):δ1.49-1.57(m,1h),1.91-1.99(m,1h),2.34-2.54(m,3h),3.34-3.38(m,1h),3.50-3.59(m,4h),3.61-3.70(m,2h),3.86(t,1h),7.21-7.24(m,2h),7.28-7.34(m,8h)。
[0056]
step 9
[0057]
将0.74g im-8和100mg钯碳溶于5ml甲醇中,氮气交换后,通氢气至30atm。加热至80℃反应48h。冷却过滤,浓缩,硅胶柱分离,得im-9纯品0.22g,收率85%。
[0058]1h nmr(400mhz,cdcl3):δ1.23(s,2h),1.47-1.52(m,1h),1.91-1.99(m,1h),2.17-2.22(m,1h),2.62-2.66(m,2h),3.40-3.45(m,1h),3.62-3.80(m,3h)。
[0059]
step 10
[0060]
将220mgim-9,284mg o-甲基-n-硝基-n
’‑
甲基异脲和83mg氯化钠放入烧瓶中,冷却至-25℃左右,加入365mg氢氧化钠水溶液(8.6%),控制温度低于-10℃,反应4h,自然升至室温后,继续反应6h,tlc检测,原料消失后停止反应,加盐酸,调节ph值小于4,反相色谱分离,得呋虫胺产品283mg,收率65%。其质谱图如图1所示。
[0061]1h nmr(400mhz,cdcl3):δ1.64-1.69(m,1h),2.10-2.14(m,1h),2.62-2.64(m,1h),2.97(s,3h),3.32-3.43(d,2h),3.62-3.66(m,1h),3.72-3.79(m,2h),3.91-3.93(m,1h)。
[0062]
实施例2
[0063]
本实施例提供一种放射性同位素碳-14标记的呋虫胺,其合成路线如下:
[0064][0065]
具体制备步骤如下:
[0066]
step 1~3同实施例1;
[0067]
step 4
[0068]
在氮气保护下,将380mg氢化锂铝分散于10ml无水四氢呋喃中,却至-5~5℃。将粗品im-3(~1.74g,10mmol)溶于10ml无水四氢呋喃中,滴加到上述溶液中。室温反应2h,回流反应2h,原料反应完全后,加水淬灭,过滤。滤饼用四氢呋喃抽提6h。浓缩,得im-4b粗品。
[0069]
step 5
[0070]
将im-4b粗品溶于甲苯中,加入2ml水和催化量的对甲苯磺酸,加热回流30min,换成分水装置。继续反应6h,液质检测,原料反应完全。浓缩,得im-5b粗品1.0g,两步总收率70%。
[0071]
step 6
[0072]
将0.8g im-5b(7.8mmol)和对甲基苯磺酰氯(1.9g,10mmol)置于干燥的反应瓶中,氮气保护下,加入20ml二氯甲烷,搅拌溶解,冷却至-5~5℃。向反应液中滴加三乙胺的二氯
甲烷溶液(1.1g,10mmol),搅拌过夜。反应完成后,反应液分别用水和饱和氯化钠溶液洗,无水硫酸钠干燥,过滤,浓缩,柱分离得im-6b纯品1.78g,收率96%。
[0073]
step 7
[0074]
将1.78g im-6b(7.6mmol)和2.28g邻苯二甲酰亚胺钾盐溶于20ml无水dmf中,加热至80℃,反应12h。tlc检测,原料反应完全后,冷却至室温。加水和乙酸乙酯,水相用乙酸乙酯萃取。合并有机相,饱和食盐水洗涤,硫酸钠干燥。过滤浓缩,硅胶柱层析分离,得im-7b产品1.6g,收率91.1%。
[0075]
step 8
[0076]
向上步制备的im-7b(1.6g,6.9mmol)的乙醇(50ml)溶液中,滴加入水合肼(1.6ml,32mmol),5min滴完。混合物回流反应90min。降温并加入20ml乙醚,减压浓缩,过滤,滤液减压脱溶得0.35g im-9,收率50%。
[0077]1h nmr(400mhz,cdcl3):δ1.23(s,2h),1.47-1.52(m,1h),1.91-1.99(m,1h),2.17-2.22(m,1h),2.62-2.66(m,2h),3.40-3.45(m,1h),3.62-3.80(m,3h)。
[0078]
step 9
[0079]
将300.5mgim-9,388.2mg o-甲基-n-硝基-n
’‑
甲基异脲(im-10)和113.2mg氯化钠放入烧瓶中,冷却至-25℃左右,加入497.8mg氢氧化钠水溶液(8.6%),控制温度低于-10℃,反应4h,自然升至室温后,继续反应6h,tlc检测,原料消失后停止反应,加盐酸,调节ph值小于4,反相色谱分离,得呋虫胺产品390.1mg,收率65.6%。
[0080]1h nmr(400mhz,cdcl3):δ1.64-1.69(m,1h),2.10-2.14(m,1h),2.62-2.64(m,1h),2.97(s,3h),3.32-3.43(d,2h),3.62-3.66(m,1h),3.72-3.79(m,2h),3.91-3.93(m,1h)。
[0081]
实施例3
[0082]
本实施例提供一种放射性同位素碳-14标记的呋虫胺,其合成路线如下:
[0083][0084]
具体制备步骤如下:
[0085]
step 1~6同实施例2;
[0086]
step7
[0087]
将0.89g im-6b(3.5mmol)和0.33g叠氮化钠溶于10ml无水dmf中,加热至80℃,反应12h。tlc检测,原料反应完全后,冷却至室温。加水和乙酸乙酯,水相用乙酸乙酯萃取。合并有机相,饱和食盐水洗涤,硫酸钠干燥。过滤浓缩,得im-7c粗品0.40g,收率90%。
[0088]
step 8
[0089]
将0.40g im-7c和50mg钯碳溶于5ml甲醇中,氮气交换后,通氢气约2个大气压,室温反应8h。过滤,浓缩,得im-9粗品0.3g,收率95%。
[0090]
step 9同实施例2。
[0091]
实施例4[14c-furanyl]-呋虫胺在产蛋鸡体内代谢实验中的应用
[0092]
试验按照《畜禽中农药代谢试验准则》开展。将[14c-furanyl]-呋虫胺与非放射性呋虫胺混合配制成比活度为50μci/mg的呋虫胺溶液,通过胶囊经口饲喂方式对10只产蛋母鸡连续给药7天。研究[14c-furanyl]-呋虫胺连续7d经口给药后产蛋母鸡中总放射性残留量(trr)、代谢产物种类及性质,并分析呋虫胺在产蛋母鸡中的代谢途径。10只母鸡各组织、粪便、蛋清和蛋黄中总放射性残留量相对含量(%trr)为90.97%。母鸡粪便和笼洗液中的%trr分别为89.66%和0.59%,在组织、血液、蛋清和蛋黄样本中的%trr在《0.01%-0.10%之间,表明呋虫胺及其代谢产物很容易通过粪便排出体外。
[0093]
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。