1.本发明涉及无机非金属材料制备与合成领域,特别涉及一种硅酸镓镧系列晶体多晶料及其制备方法。
背景技术:
2.硅酸镓镧系列晶体是一种性能优异的新型高温压电晶体材料,压电系数为4~8pc/n,具有零频率温度系数切型,机电耦合系数约为石英2~3倍,且至熔点(1300~1500℃)无相变,可广泛应用于航空航天、武器装备、汽车电子及石油化工等领域。对于晶体生长工艺而言,多晶原料的制备至关重要。传统硅酸镓镧系列晶体多晶原料的制备方法是固相反应法,通过将原料在坩埚内经1100℃固相反应12h得到硅酸镓镧多晶料,再将烧结好的多晶料放入铱金坩埚内熔化长晶,此方法能够批量制备多晶原料,但由于原料种类较多、粉体尺寸相差较大导致难以混合均匀,从而使得多组分原料在烧结时时常出现固相反应不充分、多晶料结晶性差的问题。
3.中国发明专利cn101190798a发明了一种将高纯原料溶解在柠檬酸中,加热得到粘稠凝胶,再进行烘干、煅烧得到铌酸镓镧系列纳米粉末,此法解决了多晶料组分均一性差的问题,但在制备过程中以硝酸盐作为原料,并且使用氢氟酸进行溶解,容易使产物中残留氮、氟元素,难以满足高质量晶体生长要求,同时,氢氟酸的使用不符合安全、绿色环保的生产理念。
技术实现要素:
4.针对上述硅酸镓镧多晶料制备过程中存在的问题,本发明提供一种硅酸镓镧系列晶体多晶料的制备方法,该方法简单、绿色环保。
5.实现本发明目的的技术方案是:一种硅酸镓镧系列晶体多晶料的制备方法,包括如下步骤:
6.s1:将可溶性镧镓原料溶解于有机酸溶液中,加热至充分溶解形成无色透明溶液,其中,所述可溶性镧镓原料和所述有机酸溶液的摩尔比为1:5~20;
7.s2:向上述溶液中加入难溶氧化物纳米颗粒,并搅拌、超声得到白色悬浊液,再通过球磨进一步混合;
8.s3:稀释所述白色悬浊液,并用碱性溶液调节ph至8~12,过滤得到白色滤饼;
9.s4:将所述白色滤饼洗涤、干燥得到多晶料前驱体;
10.s5:将所述多晶料前驱体进一步干燥,冷却至室温后进行压块,得到多晶料块;
11.s6:将所述多晶料块高温烧结得到硅酸镓镧系列晶体多晶料。
12.进一步地,所述硅酸镓镧系列晶体多晶料包括硅酸镓镧多晶料、铌酸镓镧多晶料、钽酸镓镧多晶料。
13.进一步地,所述可溶性镧镓原料包括镧的碳酸盐或氢氧化物以及镓的碳酸盐或氢氧化物,制备所述硅酸镓镧多晶料时,所述可溶性镧镓原料中镧和镓的摩尔比为3:5.025~
5.1;制备所述铌酸镓镧多晶料或钽酸镓镧多晶料时,所述可溶性镧镓原料中镧和镓的摩尔比为3:5.528~5.61。
14.进一步地,所述难溶氧化物纳米颗粒为二氧化硅、五氧化二钽、五氧化二铌中的一种,所述难溶氧化物纳米颗粒与所述可溶性镧镓原料的摩尔比为la:ga:si=3:5.025~5.1:1,或者la:ga:m=3:5.528~5.61:0.5,m为nb或ta。
15.进一步地,所述有机酸为柠檬酸、苹果酸、酒石酸、草酸的一种或多种。
16.进一步地,所述s3中的碱性溶液为氨水、碳酸铵、碳酸氢铵中的一种或多种。
17.进一步地,所述s4中,将所述白色滤饼洗涤具体为采用去离子水洗涤2~4次,干燥采用烘箱烘干,干燥温度为110~150℃,干燥时间为2~5h。
18.进一步地,所述s5中的多晶料前驱体进一步干燥的干燥温度为700~900℃,干燥时间为5-10h。
19.进一步地,所述s1中的加热温度为80~130℃,反应时间为1~5h。
20.进一步地,所述s2中搅拌的速率为30~90r/min,搅拌的时间为0.5~3h;所述s2中超声的频率为150~300hz,超声的时间为0.5~3h;所述s2中球磨的速率为100~300r/min,球磨的时间为5~10h。
21.进一步地,所述s6中的高温烧结的气氛为n2、ar或空气中的一种;高温烧结温度为1100~1300℃,高温烧结时间为5~12h。
22.本发明还提供一种硅酸镓镧系列晶体多晶料,所述硅酸镓镧系列晶体多晶料采用如上所述的制备方法制得。
23.本发明的有益效果在于:本发明通过将多组分原料部分溶解后再混合,利用液体易于均匀混合的特点,得到均匀的固液混合物,再通过加入碱性溶液将液相中的物质析出,得到混合均匀的固相粉末。本发明通过简单有效的办法实现了多组分原料的均匀混合,避免了传统混合过程中混合不均的问题,易于烧结得到均匀性好、结晶度高的硅酸镓镧系列晶体多晶料,且制备过程绿色环保,适合大规模制备。同时本发明在较低的温度下实现了固相反应充分完全,大大降低了晶体内部缺陷,采用本发明制备的多晶料生长的晶体的电阻率在600℃可提升至10^8ω
·
cm以上。
24.上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例并配合附图详细说明如后。
附图说明
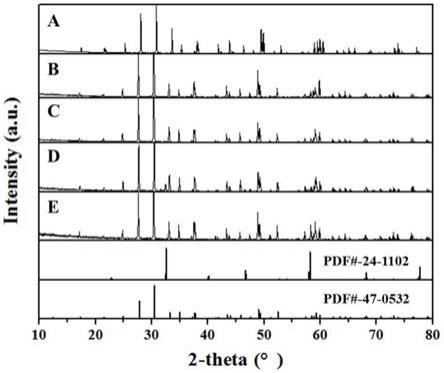
25.图1为请本发明实施例1-5中所制备样品的xrd图,其中,a为实施例1样品,b为实施例2样品,c为实施例3样品,d为实施例4样品,e为实施例5样品。
26.图2本发明实施例1-5中所制备样品的sem图,其中,a为实施例1样品,b为实施例2样品,c为实施例3样品,d为实施例4样品,e为实施例5样品。
具体实施方式
27.下面将结合附图对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
28.此外,下面所描述的本发明不同实施方式中所涉及的技术特征只要彼此之间未构成冲突就可以相互结合。
29.本发明提供硅酸镓镧系列晶体多晶料的制备方法,硅酸镓镧系列晶体多晶料包括硅酸镓镧(la3ga5sio
14
)多晶料、铌酸镓镧(la3ga
5.5
nb
0.5o14
)多晶料、钽酸镓镧(la3ga
5.5
ta
0.5o14
)多晶料。
30.该制备方法包括如下步骤:
31.s1:将可溶性镧镓原料溶解于有机酸溶液中,加热至充分溶解形成无色透明溶液,其中,可溶性镧镓原料和有机酸溶液的摩尔比为1:5~20;
32.s2:向上述溶液中加入难溶氧化物纳米颗粒,并搅拌、超声得到白色悬浊液,再通过球磨进一步混合;
33.s3:稀释白色悬浊液,并用碱性溶液调节ph至8~12,过滤得到白色滤饼;
34.s4:将白色滤饼洗涤、干燥得到多晶料前驱体;
35.s5:将多晶料前驱体进一步干燥,冷却至室温后进行压块,得到多晶料块;
36.s6:将多晶料块高温烧结得到硅酸镓镧系列晶体多晶料。
37.其中,可溶性镧镓原料包括镧的碳酸盐或氢氧化物以及镓的碳酸盐或氢氧化物,制备硅酸镓镧多晶料时,可溶性镧镓原料中镧和镓的摩尔比为3:5.025~5.1;制备铌酸镓镧多晶料或钽酸镓镧多晶料时,可溶性镧镓原料中镧和镓的摩尔比为3:5.528~5.61。
38.难溶氧化物纳米颗粒为二氧化硅、五氧化二钽、五氧化二铌中的一种,难溶氧化物纳米颗粒与可溶性镧镓原料的摩尔比为la:ga:si=3:5.025~5.1:1,或者la:ga:m=3:5.528~5.61:0.5,m为nb或ta。
39.有机酸为柠檬酸、苹果酸、酒石酸、草酸的一种或多种。有机酸的使用量相对可溶性镧镓原料过量,保证可溶性镧镓原料充分溶解,形成透明溶液。
40.s3中的碱性溶液为氨水、碳酸铵、碳酸氢铵中的一种或多种。有机酸和碱性溶液还可以为其他,在此不做具体限定。
41.s1中的加热温度为80~130℃,反应时间为1~5h。
42.s2中搅拌的速率为30~90r/min,搅拌的时间为0.5~3h;s2中超声的频率为150~300hz,超声的时间为0.5~3h;s2中球磨的速率为100~300r/min,球磨的时间为5~10h。
43.s4中,将白色滤饼洗涤具体为采用去离子水洗涤2~4次,干燥采用烘箱烘干,干燥温度为110~150℃,干燥时间为2~5h。
44.s5中的多晶料前驱体进一步干燥的干燥温度为700~900℃,干燥时间为5-10h。
45.s6中的高温烧结的气氛为n2、ar或空气中的一种;高温烧结温度为1100~1300℃,高温烧结时间为5~12h。
46.下面以具体实施例对上述制备方法进行详细介绍。
47.(实施例1)
48.按摩尔比la:ga:si=3:5.05:1称取氢氧化镧、氢氧化镓、二氧化硅纳米颗粒,将氢氧化镧、氢氧化镓可溶性原料加入到1.5mol/l的柠檬酸溶液中,可溶性原料与柠檬酸的摩尔比为1:8,100℃加热反应2.5h至液体呈无色透明,再加入二氧化硅纳米颗粒以30r/min的速率搅拌3h,150hz的频率超声3h得到白色悬浊液。将白色悬浊液加入到球磨仪内球磨混合,球磨速率200r/min,球磨时间8h。向球磨后的白色悬浊液中加去离子水稀释,接着加入
适量氨水调节白色悬浊液的ph为11。将白色悬浊液过滤洗涤,所得滤饼放置于110℃烘箱中烘干5h得到多晶料前驱体,再经850℃预烧结5h,冷却至室温后进行压块,在空气中1300℃烧结5h得到硅酸镓镧多晶料。请参见图1和图2,所制备的样品进行xrd和sem表征,结果表明样品为纯相结构,结晶性较好,样品颗粒尺寸在10~30nm之间,颗粒均匀性较好。
49.(实施例2)
50.按摩尔比la:ga:nb=3:5.555:0.5称取氢氧化镧、氢氧化镓、五氧化二铌纳米颗粒,将氢氧化镧、氢氧化镓可溶性原料加入到1.5mol/l的柠檬酸溶液中,可溶性原料与柠檬酸的摩尔比为1:8,100℃加热反应2.5h至液体呈无色透明,再加入五氧化二铌纳米颗粒以30r/min的速率搅拌3h,150hz的频率超声3h得到白色悬浊液。将白色悬浊液加入到球磨仪内球磨混合,球磨速率150r/min,球磨时间10h。向球磨后的白色悬浊液中加去离子水稀释,接着加入适量氨水调节白色悬浊液的ph为11。将白色悬浊液过滤洗涤,所得滤饼放置于110℃烘箱中烘干5h得到多晶料前驱体,再经850℃预烧结5h,冷却至室温后进行压块,在空气中1300℃烧结5h得到铌酸镓镧多晶料。请参见图1和图2,所制备的样品进行xrd和sem表征,结果表明样品为纯相结构,结晶性较好,样品颗粒尺寸在10~30nm之间,颗粒均匀性较好。
51.(实施例3)
52.按摩尔比la:ga:ta=3:5.555:0.5称取碳酸镧、碳酸镓、五氧化二钽纳米颗粒,将碳酸镧、碳酸镓可溶性原料加入到1.5mol/l的柠檬酸溶液中,可溶性原料与柠檬酸的摩尔比为1:16,100℃加热反应2.5h至液体呈无色透明,再加入五氧化二钽纳米颗粒以30r/min的速率搅拌3h,150hz的频率超声3h得到白色悬浊液。将白色悬浊液加入到球磨仪内球磨混合,球磨速率250r/min,球磨时间6h。向球磨后的白色悬浊液中加去离子水稀释,接着加入适量氨水调节白色悬浊液的ph为11。将白色悬浊液过滤洗涤,所得滤饼放置于110℃烘箱中烘干5h得到多晶料前驱体,再经850℃预烧结5h,冷却至室温后进行压块,在空气中1300℃烧结5h得到钽酸镓镧多晶料。请参见图1和图2,所制备的样品进行xrd和sem表征,结果表明样品为纯相结构,结晶性较好,样品颗粒尺寸在10~30nm之间,颗粒均匀性较好。
53.(实施例4)
54.按摩尔比la:ga:ta=3:5.555:0.5称取氢氧化镧、氢氧化镓、五氧化二钽纳米颗粒,将氢氧化镧、氢氧化镓可溶性原料加入到1.5mol/l的柠檬酸溶液中,可溶性原料与柠檬酸的摩尔比为1:8,100℃加热反应2.5h至液体呈无色透明,再加入五氧化二钽纳米颗粒以30r/min的速率搅拌3h,150hz的频率超声3h得到白色悬浊液。将白色悬浊液加入到球磨仪内球磨混合,球磨速率200r/min,球磨时间8h。向球磨后的白色悬浊液中加去离子水稀释,接着加入适量氨水调节白色悬浊液的ph为9。将白色悬浊液过滤洗涤,所得滤饼放置于110℃烘箱中烘干5h得到多晶料前驱体,再经850℃预烧结5h,冷却至室温后进行压块,在n2气氛中1300℃烧结5h得到钽酸镓镧多晶料。
55.请参见图1和图2,所制备的样品进行xrd和sem表征,结果表明样品基本为纯相结构,样品颗粒尺寸在5~20nm之间。
56.(实施例5)
57.按摩尔比la:ga:ta=3:5.555:0.5称取氢氧化镧、氢氧化镓、五氧化二钽纳米颗粒,将氢氧化镧、氢氧化镓可溶性原料加入到1.5mol/l的柠檬酸溶液中,可溶性原料与柠檬酸的摩尔比为1:8,100℃加热反应2.5h至液体呈无色透明,再加入五氧化二钽纳米颗粒以
30r/min的速率搅拌3h,150hz的频率超声3h得到白色悬浊液。将白色悬浊液加入到球磨仪内球磨混合,球磨速率200r/min,球磨时间8h。向球磨后的白色悬浊液中加去离子水稀释,接着加入适量氨水调节白色悬浊液的ph为12。将白色悬浊液过滤洗涤,所得滤饼放置于110℃烘箱中烘干5h得到多晶料前驱体,再经850℃预烧结5h,冷却至室温后进行压块,ar气氛中1300℃烧结5h得到钽酸镓镧多晶料。
58.请参见图1和图2,所制备的样品进行xrd和sem表征,结果表明样品为纯相结构,结晶性较好,样品颗粒尺寸在10~30nm之间,颗粒均匀性较好。
59.本发明通过将多组分原料部分溶解后再混合,利用液体易于均匀混合的特点,得到均匀的固液混合物,再通过加入碱性溶液将液相中的物质析出,得到混合均匀的固相粉末。本发明通过简单有效的办法实现了多组分原料的均匀混合,避免了传统混合过程中混合不均的问题,易于烧结得到均匀性好、结晶度高的硅酸镓镧系列晶体多晶料,且制备过程绿色环保,适合大规模制备。
60.以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
61.以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。