shen,org.process res.dev.2019,23,852-857;张耀春,王兵,院兴,罗绪,左小勇,雷皇书,重庆医药工业研究院有限责任公司,cn104844585a,2015-04-15;王雪根,何凌云,余洋,金皓杰,郭莉芹,南京艾德凯腾生物医药言笑自若有限责任公司,cn105461704a,2015-12-15;公绪栋,陈健,郭玉辉,应述欢,上海博志研新药物技术有限公司,cn107674067a,2017-10-19;孙长亮,胡天文,陈伟铭,何洋,蒋翔锐,上海特化医药科技有限公司,cn109307716 a,2017-7-27)。并且对于该合成路线所产生的杂质进行了详细的研究(rahul tyagi,harnam singh,jagat singh,himanshu arora,vijayalaxmi yelmeli,mohit jain,sathyanarayana girigani,and pramod kumar,org.process res.dev.2018,22,1471-1480;罗绪,付廷印,张耀春,张华娇,左小勇,雷皇书,邓杰,重庆医药工业研究院有限责任公司,cn106892909a,2015-12-18;代广会,陈志锋,万娟,张道林,重庆医药工业研究院有限责任公司,cn107525877a,2016-6-20;柯潇,张伟,成都弘达药业有限公司,cn109307716 a,2017-7-27;柯潇,杨俊,成都弘达药业有限公司,cn111909159 a,2019-5-8;柯潇,张伟,成都弘达药业有限公司,cn111912914 a,2019-5-8)。
技术实现要素:
8.有鉴于此,本技术首先提供一种依匹哌唑的制备方法,反应物料使用1-叔丁氧甲酰-7-(4-氯-丁氧)-喹啉-2-酮,规避了7-(4-氯-丁氧)-1h-喹啉-2-酮自身引起的分子内和分子间的缩合反应,反应选择性高,产品质量优,适合工业化生产。
9.具体地,本技术是通过以下方案实现的:
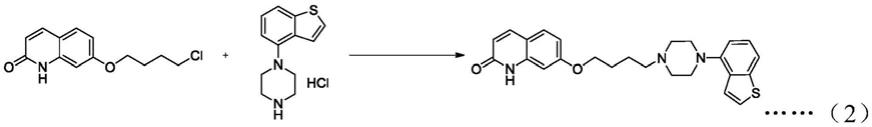
10.一种依匹哌唑合成方法,以4-哌嗪-苯并噻吩盐酸盐、1-叔丁氧甲酰-7-(4-卤素-丁氧)-喹啉-2-酮为起始原料,碳酸钾为缚酸剂,在乙醇-水混合溶液中回流反应合成叔丁氧甲酰保护的依匹哌唑。经过乙醇-乙酸混合溶液中高温下与浓盐酸反应脱除叔丁氧甲酰保护基并形成依匹哌唑盐酸盐,所得依匹哌唑盐酸盐在乙醇-乙酸溶液回流搅洗后,乙醇-水混合溶液中与氢氧化钠溶液中和得到依匹哌唑,去离子水洗涤,干燥得到依匹哌唑成品。
11.上述合成方法的收率约50%,成品经hplc检测纯度大于99.5%,最大单杂小于0.1%。与以往的亲核取代反应合成依匹哌唑相比,反应物料使用1-叔丁氧甲酰-7-(4-氯-丁氧)-喹啉-2-酮,规避了7-(4-氯-丁氧)-1h-喹啉-2-酮自身引起的分子内和分子间的缩合反应,反应选择性高,产品质量优,适合工业化生产。
12.其中上述过程中,合成的依匹哌唑为上述式(ⅰ)所示结构,起始原料4-哌嗪-苯并噻吩盐酸盐的结构如式(3)所示,1-叔丁氧甲酰-7-(4-卤素-丁氧)-喹啉-2-酮的结构如式(4)所示。
13.14.其中:x=br,cl
…………………………
式(4)
15.上述过程以乙醇-水为溶剂,在碳酸钾为缚酸剂条件下进行亲核取代反应,经过脱除叔丁氧甲酰保护基并形成依匹哌唑盐酸盐,氢氧化钠中和得到式(1)化合物,其反应过程可如式(5)所示。
[0016][0017]
进一步的,作为优选:
[0018]
所示1-叔丁氧甲酰-7-(4-卤素-丁氧)-喹啉-2-酮是由7-(4-卤素-丁氧)-1h-喹啉-2-酮与叔丁氧甲酸酐(boc2o)制备,制备过程如式(6)所示:
[0019][0020]
所述1-叔丁氧甲酰-7-(4-卤素-丁氧)-喹啉-2-酮的卤素选自氯、溴,更优选的,所述1-叔丁氧甲酰-7-(4-卤素-丁氧)-喹啉-2-酮为1-叔丁氧甲酰-7-(4-氯-丁氧)-喹啉-2-酮。
[0021]
所述溶剂为乙醇-水为溶剂,乙醇-水的配比为0.2-2(体积比),优选0.4。
[0022]
所述1-叔丁氧甲酰-7-(4-卤素-丁氧)-喹啉-2-酮:4-哌嗪-苯并噻吩盐酸盐的摩尔比为0.8:1-1:1.2,优选1:1。
[0023]
所述反应温度优选60-90℃,更优选75-80℃。
[0024]
我司在开发依匹哌唑原料药过程中,参考了已发表的文献和公开的专利,决定采用中科院上海药物所沈敬山研究员发表的合成方法(weiming chen,jin suo,yongjian liu,yuanchao xie,mingjun wu,fuqiang zhu,yifeng nian,haji a.aisa,and jingshan shen,org.process res.dev.2019,23,852-857),在重复该合成路线时,合成依匹哌唑难以得到文献描述的高质量,最终产品依匹哌唑经hplc分析有一个杂质含量高于0.1%,难以用普通的乙醇-水体系,二氯甲烷-甲醇-甲基叔丁基醚体系,二氯甲烷-甲醇-乙酸乙酯体系重结晶去除该杂质,从反应的原料分析很可能是7-(4-氯-丁氧)-1h-喹啉-2-酮不稳定,发生了分子内或者分子间自身缩合所引起的副反应产生的杂质(韩峰,王婷,冯斌,徐清,有机化学,2021,41,2831~2838),具体见式(7)的7-(4-氯-丁氧)-1h-喹啉-2-酮分子内自身缩合反应和式(8)的7-(4-氯-丁氧)-1h-喹啉-2-酮分子间自身缩合。
[0025][0026]
基于以上推断,为了减少7-(4-氯-丁氧)-1h-喹啉-2-酮所引起的分子内或者分子间自身缩合副反应发生,我们设计用叔丁氧甲酸酐(boc2o)保护1位n原子,所得物料再与4-哌嗪-苯并噻吩盐酸盐缩合来合成依匹哌唑,取得良好效果,见式(9)、(10)。
[0027][0028][0029]
本发明上述合成方法工艺简单、反应条件温和、反应选择性高,产品质量优,适合工业化生产的依匹哌唑制备方法。
附图说明
[0030]
图1为实施例1合成中间体的hplc检测图谱;
[0031]
图2为实施例2合成中间体的hplc检测图谱;
[0032]
图3为实施例3合成中间体的hplc检测图谱;
[0033]
图4为实施例3合成依匹哌唑中间体的1h谱图;
[0034]
图5为实施例3合成依匹哌唑中间体c
13
谱图;
[0035]
图6为成品依匹哌唑的hplc检测图谱。
具体实施方式
[0036]
实施例1:合成7-(4-氯丁氧基)-3,4-二氢-2(1h)-喹啉酮
[0037]
将7-羟基-3,4-二氢-2(1h)-喹啉酮(200g,1.23mol)、1-溴-4-氯丁烷(631g,3.68mol)以及碳酸钾(254g,1.84mol)在dmf(800ml)投入5l三口烧瓶中混合搅拌,然后25℃搅拌30h,悬浊液逐渐越来越稠厚。将纯化水(1600ml)在搅拌下缓慢倒入到上述5l三口烧瓶中,析出大量固体,继续搅拌30min后抽滤,并用纯化水进行洗涤固体。将上述所得固体转移到1l三口烧瓶中,加入甲基叔丁基醚(400ml)中打浆搅洗1h,然后抽滤,所得固体50℃真空干燥,得灰白色固体(258g),收率约83.6%。hplc检测(见图1)纯度约96.14%(其中7-(4-溴丁氧基)-3,4-二氢-2(1h)-喹啉酮含量1.76%)。
[0038]
表1:本实施例中间体的hplc测试结果数据摘录
[0039][0040]
实施例2:合成7-(4-氯丁氧基)-喹啉-2-酮
[0041]
将7-(4-氯丁氧基)-3,4-二氢-2(1h)-喹啉酮(150g,0.59mol),thf(750ml)投入1l三口烧瓶中,室温搅拌然后缓慢分批加入ddq(161g,0.71mol),继续搅拌18h。将上述反应液缓慢倒入水(750ml)中进行淬灭,然后在搅拌下缓慢加入碳酸氢钠水溶液(1.57mol/l,750ml),搅拌1h。抽滤,所得固体用水洗涤,50℃真空干燥,得到淡黄色固体(122g),收率为82.1%。hplc检测(见图2)纯度约97.43%(其中7-(4-溴丁氧基)-喹啉酮含量1.67%)。
[0042]
表2:本实施例中间体的hplc测试结果数据摘录
[0043][0044]
实施例3:合成1-n-叔丁氧甲酰-7-(4-氯丁氧基)-喹啉-2-酮
[0045]
将7-(4-氯丁氧基)-喹啉-2-酮(40.2g,160mmol)以及dmap(1.9g,16mmol)在二氯甲烷(200ml)中混合,室温搅拌下滴加二碳酸二叔丁酯(70.0g,320mmol),二氯甲烷(100ml)溶液,随后35℃反应1h。将上述二氯甲烷反应液使用水(100ml x 3)洗涤,然后采用无水硫酸钠干燥。过滤,然后在35℃下减压蒸馏除去二氯甲烷,所得固体并加入甲基叔丁基醚(400ml)搅洗1h。过滤,并用甲基叔丁基醚洗涤固体,真空干燥,得到白色固体(44.9g),产率约为79.8%。hplc检测(见图3)纯度约97.88%(其中1-n-叔丁氧甲酰-7-(4-溴丁氧基)-喹啉-2-酮含量1.55%)。
[0046]
表3:本实施例中间体的hplc测试结果数据摘录
[0047][0048]
esi-ms:m/z=352.00[m+h].(见图4、图5)
[0049]
1h-nmr(500mhz,dmso-d6)δ(ppm):8.41(d,j=8.2hz,1h),7.92(d,j=9.0hz,1h),7.32(d,j=2.5hz,1h),7.27(t,j=2.5hz,1h),7,19(t,j=8.5hz,1h),4.17(dd,j=5.5hz,2h),3.73(t,j=2.5hz,2h),2.50(t,j=2.0hz,2h),1.91(t,j=2.5hz,2h),1.52(s,9h).
[0050]
13c-nmr(125mhz,dmso-d6)δ(ppm):160.74,156.77,150.99,148.23,140.85,129.50,122.38,119.76,113.02,107.86,84.25,67.68,45.60,29.35,27.72,26.43.
[0051]
实施例4:合成依匹哌唑
[0052]
将4-哌嗪苯并噻吩盐酸盐(10.7g,42mmol)以及碳酸钾(5.8g,42mmol)在乙醇-水混合液(乙醇:水=1:2.5,25ml)中混合,室温搅拌下通氮气鼓泡60min,升温至90℃分批加入1-n-叔丁氧甲酰-7-(4-氯丁氧基)-喹啉-2-酮(14.1g,40mmol),保温18h后冷却至室温,过滤,所得固体真空干燥,得到棕色固体(约16.9g),。将上述棕色固体加入乙醇(340ml)和乙酸(21ml)中分散,然后机械搅拌升温至80℃,缓慢滴加浓盐酸(~35%,3.6ml,34mmol),搅拌1h,然后冷却至20℃以下,抽滤,所得固体用乙醇洗涤,然后真空干燥,得到淡黄色依匹哌唑盐酸盐(约13.4g)。将上述所得依匹哌唑盐酸盐分散在乙醇-水混合液(乙醇:水=1.5:1,188ml)中,升温至80℃搅拌溶解,然后加入活性炭(1.4g),回流1h,趁热抽滤,然后降温至20℃以下析晶,得到本白色依匹哌唑盐酸盐(约10.8g)。将上述所得依匹哌唑盐酸盐粗品(10.8g,)分散在乙醇-水混合液(乙醇:水=1.5:1,188ml)中,升温至80℃搅拌溶解,然后滴加氢氧化钠水溶液(20%,5.5g,28mmol),回流1h,然后降温至20~25℃析晶,抽滤,所得并用水洗涤,真空干燥,得到白色依匹哌唑固体(8.9g),产率为51.3%。hplc检测(见图6)纯度约99.77%,最大单杂0.058%(小于0.1%)。
[0053]
表4:本实施例成品的hplc测试结果数据摘录
[0054][0055]
hplc检测色谱条件:
[0056]
色谱柱:zorbaxeclipseplusc185um4.6*250mm
[0057]
流动相a:0.01m磷酸二氢钾用氨水调节ph至7.00
[0058]
流动相b:甲醇:乙腈=20:80。
[0059]
梯度洗脱程序如表1所示。
[0060]
表1:梯度洗脱程序参数表
[0061][0062][0063]
溶剂:甲醇:乙腈=20:80
[0064]
检测波长:225nm柱温:25℃
[0065]
流速:1.0ml/min进样量:20μl。