1.本发明属于半导体材料领域,具体涉及一种含可自交联巯基三聚氰胺聚合物的抗反射涂层组合物及其制备方法和图案形成方法。
背景技术:
2.在半导体制造中,一直以来都是通过对光致抗蚀剂的光刻来实现细微加工。光致抗蚀剂也称为光刻胶,被用来将图像转移到基底上。在基底上形成光致抗蚀剂的涂层后,通过光掩模将光致抗蚀剂层曝光于活化辐射源。在曝光之后,光致抗蚀剂层上会发生化学修饰反应,光掩模就有光辐射透过和不透过区域,因此光掩模的图案被转移到光刻抗蚀剂涂层上。之后,光刻抗蚀剂涂层被显影形成能够在基底上被选择性处理的图案化图像。
3.在曝光工艺中,当辐射到的光致抗蚀剂涂层的光辐射被反射时,在光致抗蚀剂涂层上的图案化的图像分辨率就会降低。例如,当光辐射在基底和光致抗蚀剂之间的界面上被反射时,引起了辐照到光致抗蚀剂涂层上的光化辐射强度的空间变化,且光化辐射向着非预想的光致抗蚀剂区域散射,引起显影后图案的线宽变化或缺乏均一性。另外,由于在区域间的散射光化辐射或反射光化辐射的量不同,线宽会变得不均一,例如,分辨率会由于基底的表面形貌受到限制。
4.在光刻胶下层设置底部抗反射膜(barc)是解决上述问题的最佳选择。底部抗反射膜是指在光刻胶和基体之间加入一层能有效消除光反射形成干涉驻波的底部抗反射膜。该底部抗反射膜能够增加曝光能量范围和焦距,降低基体几何结构差异对关键尺寸均匀度的影响,同时减少反射光的散射造成的圆形缺口,缓解基体构型导致光刻胶厚度不同而引起的摆动曲线效应和凹缺效应。通常,将底部抗反射涂层组合物涂覆到基材上,然后将光致抗蚀剂组合物涂覆在抗反射涂层上面。将抗反射涂层烘烤固化以防止抗反射涂层和光致抗蚀剂之间的掺混。将光致抗蚀剂成像式曝光并显影从而将光致抗蚀剂图案转印至基材上。
5.异氰尿酸酯类化合物常被用作抗反射涂层组合物的成膜物质,倘若异氰尿酸酯类化合物中含有巯基时,通常需要对巯基进行封端,以提高抗反射涂层组合物的稳定性。此外,现有的以异氰尿酸酯类化合物作为成膜物质的底部抗反射涂层组合物虽然具有较高的折射率和消光系数,但是刻蚀速率却不高。
技术实现要素:
6.本发明的目的是为了克服现有的以异氰尿酸酯类化合物作为成膜物质的底部抗反射涂层组合物无法兼具高折射率、消光系数和刻蚀速率的缺陷,而提供一种同时兼具有高折射率、消光系数和刻蚀速率的抗反射涂层组合物及其制备方法和图案形成方法。
7.具体地,本发明提供了一种抗反射涂层组合物,其中,所述抗反射涂层组合物中含有可自交联巯基三聚氰胺聚合物、酸产生剂、有机溶剂以及任选的表面活性剂,所述可自交联巯基三聚氰胺聚合物为具有式(1)所示重复单元的聚合物;
[0008][0009][0010][0011]
r1具有式(2)或式(3)所示的结构;r2、r4和r6各自独立地为氢、任选经取代的c1~c20烷基、任选经取代的c1~c20烷氧基、任选经取代的c1~c20杂烷基、任选经取代的c6~c20环烷基、任选经取代的c1~c20烷基醇、任选经取代的c6~c
20
碳环芳基或任选经取代的c6~c
20
杂芳烷基;r3和r5各自独立地为任选经取代的c1~c20烷基、任选经取代的c1~c20杂烷基、任选经取代的c6~c20环烷基、任选经取代的c6~c
20
碳环芳基或任选经取代的c6~c
20
杂芳烷基。
[0012]
在一种优选实施方式中,r2、r4和r6各自独立地为任选经取代的c1~c5烷基、任选经取代的c1~c5烷氧基、任选经取代的c6~c20环烷基、任选经取代的c1~c5烷基醇或任选经取代的c6~c
20
碳环芳基;r3和r5各自独立地为任选经取代的c1~c5烷基、任选经取代的c6~c20环烷基或任选经取代的c6~c
20
碳环芳基。
[0013]
在一种优选实施方式中,所述可自交联巯基三聚氰胺聚合物按照包括以下步骤的方法制备得到:
[0014]
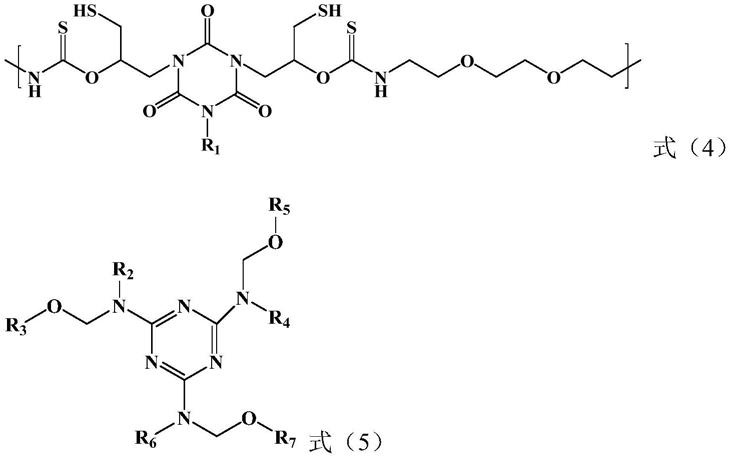
s1、将三(1,3-氧硫杂环戊烷-2-亚硫酰-5-甲基)异氰脲酸酯和1,8-二氨基-3,6-二氧杂辛烷进行开环缩聚反应,得到具有式(4)所示重复单元的预聚物;
[0015]
s2、将所述预聚物与式(5)所示的烷氧基三聚氰胺进行消除反应,得到具有式(1)所示重复单元的可自交联巯基三聚氰胺聚合物;
[0016]
[0017][0018]
r1具有式(2)或式(3)所示的结构;r2、r4和r6各自独立地为氢、任选经取代的c1~c20烷基、任选经取代的c1~c20烷氧基、任选经取代的c1~c20杂烷基、任选经取代的c6~c20环烷基、任选经取代的c1~c20烷基醇、任选经取代的c6~c
20
碳环芳基或任选经取代的c6~c
20
杂芳烷基,优选各自独立地为任选经取代的c1~c5烷基、任选经取代的c1~c5烷氧基、任选经取代的c6~c20环烷基、任选经取代的c1~c5烷基醇或任选经取代的c6~c
20
碳环芳基;r3、r5和r7各自独立地为任选经取代的c1~c20烷基、任选经取代的c1~c20杂烷基、任选经取代的c6~c20环烷基、任选经取代的c6~c
20
碳环芳基或任选经取代的c6~c
20
杂芳烷基,优选各自独立地为任选经取代的c1~c5烷基、任选经取代的c6~c20环烷基或任选经取代的c6~c
20
碳环芳基。
[0019]
在一种优选实施方式中,步骤s1中,所述三(1,3-氧硫杂环戊烷-2-亚硫酰-5-甲基)异氰脲酸酯与1,8-二氨基-3,6-二氧杂辛烷的摩尔比为1:(2~2.5)。
[0020]
在一种优选实施方式中,步骤s1中,所述开环缩聚反应的条件包括温度为室温,时间为20~30h。
[0021]
在一种优选实施方式中,步骤s2中,所述三(1,3-氧硫杂环戊烷-2-亚硫酰-5-甲基)异氰脲酸酯与烷氧基三聚氰胺的摩尔比为1:(0.8~1.2)。
[0022]
在一种优选实施方式中,步骤s2中,所述消去反应的条件包括温度为40~50℃,时间为1~5h。
[0023]
在一种优选实施方式中,所述烷氧基三聚氰胺选自以下化合物中的至少一种:
[0024]
[0025][0026]
在一种优选实施方式中,所述可自交联巯基三聚氰胺聚合物的重均分子量为5000~15000,优选为6000~14000,更优选为7000~10000。
[0027]
在一种优选实施方式中,所述可自交联巯基三聚氰胺聚合物的含量为0.5~8wt%,所述酸产生剂的含量为0.01~1wt%,所述有机溶剂的含量为90~99wt%,所述表面活性剂的含量为0.001~1wt%。
[0028]
在一种优选实施方式中,所述酸产生剂为光酸发生剂和/或热酸发生剂;所述光酸发生剂选自十二烷基苯磺酸、对甲苯磺酸、苯二甲酰亚氨基三氟甲磺酸酯、苯二甲酰亚氨基三氟甲磺酸盐、二硝基苄基甲苯磺酸酯、二硝基苄基甲苯磺酸盐、正癸基二砜、萘基亚氨基三氟甲磺酸酯、萘基亚氨基三氟甲磺酸盐、二苯基碘三氟甲磺酸盐、二苯基碘全氟丁基磺酸盐、二苯基碘六氟磷酸盐、二苯基碘六氟砷酸盐、二苯基碘六氟锑酸盐、二苯基对甲氧基苯基锍三氟甲磺酸盐、二苯基对甲苯锍三氟甲磺酸盐、二苯基对叔丁基苯基锍三氟甲磺酸盐、二苯基对异丁基苯基锍三氟甲磺酸盐、三苯基锍三氟甲磺酸盐、三(对叔丁基苯基)锍三氟甲磺酸盐、二苯基对甲氧基苯基锍全氟丁基磺酸盐、二苯基对甲苯基锍全氟丁基磺酸盐、二苯基对叔丁基苯基锍全氟丁基磺酸盐、二苯基对异丁基苯基锍全氟丁基磺酸盐、三苯基锍全氟丁基磺酸盐、三对叔丁基苯基锍全氟丁基磺酸盐、六氟砷酸酯、六氟砷酸盐、三苯基锍六氟锑酸盐和二丁基萘基锍三氟甲磺酸盐中的至少一种。
[0029]
在一种优选实施方式中,所述有机溶剂选自2-羟基异丁酸甲酯、环己酮、环戊酮、丁内酯、二甲基乙酰胺、二甲基甲酰胺、二甲基亚砜、n-甲基吡咯烷酮、四氢糠醇、丙二醇单
甲醚、丙二醇单甲醚乙酸酯和乳酸乙酯中的至少一种。
[0030]
在一种优选实施方式中,所述表面活性剂为氟化表面活性剂和/或非氟化表面活性剂。
[0031]
本发明还提供了所述抗反射涂层组合物的制备方法,该方法包括将可自交联巯基三聚氰胺聚合物、酸产生剂、有机溶剂以及任选的表面活性剂混合均匀。
[0032]
本发明还提供了一种图案形成方法,其中,该方法包括以下步骤:
[0033]
将所述抗反射涂层组合物涂敷至基底上并进行热固化,以在基底上形成抗反射涂层;
[0034]
在抗反射涂层上形成光致抗蚀剂层;
[0035]
将所述光致抗蚀剂层曝光并显影以形成光刻胶图案。
[0036]
本发明在异氰尿酸酯主链上通过巯基接三聚氰胺类化合物形成的可自交联聚合物,并且严格控制三聚氰胺类化合物的接枝率以保证聚合物上还含有适量的活性巯基,后续使用时通过活性巯基作为自交联点,如此不仅能够确保其作为成膜物质使用时无需额外添加交联剂,涂层性能稳定,更为重要的是,以这种特定结构的自交联聚合物作为成膜物质所得抗反射涂层组合物经固化后形成的涂层兼具有高折射率、消光系数和刻蚀速率,由此弥补通过端羟基连接甘脲类交联剂所形成的聚合物所存在的弊端。
具体实施方式
[0037]
本发明提供的抗反射涂层组合物中含有可自交联巯基三聚氰胺聚合物、酸产生剂和有机溶剂,优选还含有表面活性剂。其中,所述可自交联巯基三聚氰胺聚合物的含量优选为0.5~8wt%,如0.5wt%、1wt%、1.5wt%、2wt%、2.5wt%、3wt%、3.5wt%、4wt%、4.5wt%、5wt%、5.5wt%、6wt%、6.5wt%、7wt%、7.5wt%、8wt%。所述酸产生剂的含量优选为0.01~1wt%,如0.01wt%、0.05wt%、0.1wt%、0.2wt%、0.3wt%、0.4wt%、0.5wt%、0.6wt%、0.7wt%、0.8wt%、0.9wt%、1.0wt%。所述有机溶剂的含量优选为90~99wt%,如90wt%、91wt%、92wt%、93wt%、94wt%、95wt%、96wt%、97wt%、98wt%、99wt%。所述表面活性剂的含量为0~1wt%,如0、0.001wt%、0.0015wt%、0.002wt%、0.005wt%、0.008wt%、0.01wt%、0.015wt%、0.02wt%、0.05wt%、0.1wt%、0.2wt%、0.3wt%、0.4wt%、0.5wt%、0.6wt%、0.7wt%、0.8wt%、0.9wt%、1.0wt%。
[0038]
所述可自交联巯基三聚氰胺聚合物具有式(1)表示的重复单元;
[0039][0040][0041][0042]
r1具有式(2)或式(3)所示的结构;r2、r4和r6各自独立地为氢、任选经取代的c1~c20烷基、任选经取代的c1~c20烷氧基、任选经取代的c1~c20杂烷基、任选经取代的c6~c20环烷基、任选经取代的c1~c20烷基醇、任选经取代的c6~c
20
碳环芳基或任选经取代的c6~c
20
杂芳烷基,优选各自独立地为任选经取代的c1~c5烷基、任选经取代的c1~c5烷氧基、任选经取代的c6~c20环烷基、任选经取代的c1~c5烷基醇或任选经取代的c6~c
20
碳环芳基;r3和r5各自独立地为任选经取代的c1~c20烷基、任选经取代的c1~c20杂烷基、任选经取代的c6~c20环烷基、任选经取代的c6~c
20
碳环芳基或任选经取代的c6~c
20
杂芳烷基,优选各自独立地为任选经取代的c1~c5烷基、任选经取代的c6~c20环烷基或任选经取代的c6~c
20
碳环芳基。其中,所述任选经取代的c1~c5烷基的具体实例包括但不限于:甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、异戊基或新戊基。所述任选经取代的c1~c5烷氧基的具体实例包括但不限于:甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基、正戊氧基、异戊氧基或新戊氧基。所述任选经取代的c6~c20环烷基的具体实例包括但不限于:环己基、甲基环己基、乙基环己基或丁基环己基。所述任选经取代的c1~c5烷基醇的具体实例包括但不限于:甲醇基、乙醇基、正丙醇基、异丙醇基、正丁醇基、异丁醇基、叔丁醇基、正戊醇基、异戊醇基或新戊醇基。所述任选经取代的c6~c
20
碳环芳基的具体实例包括但不限于:苯基、甲苯基或苯甲基。
[0043]
在一种优选实施方式中,所述可自交联巯基三聚氰胺聚合物按照包括以下步骤的方法制备得到:
[0044]
s1、将三(1,3-氧硫杂环戊烷-2-亚硫酰-5-甲基)异氰脲酸酯和1,8-二氨基-3,6-二氧杂辛烷进行开环缩聚反应,反应过程如方程式(ⅰ)所示,得到预聚物;
[0045]
s2、将所述预聚物与烷氧基三聚氰胺进行消除反应,反应过程如方程式(ⅱ)所示,
得到可自交联巯基三聚氰胺聚合物;
[0046][0047][0048]
r1具有式(2)或式(3)所示的结构;r2、r4和r6各自独立地为氢、任选经取代的c1~c20烷基、任选经取代的c1~c20烷氧基、任选经取代的c1~c20杂烷基、任选经取代的c6~c20环烷基、任选经取代的c1~c20烷基醇、任选经取代的c6~c
20
碳环芳基或任选经取代的c6~c
20
杂芳烷基,优选各自独立地为任选经取代的c1~c5烷基、任选经取代的c1~c5烷氧基、任选经取代的c6~c20环烷基、任选经取代的c1~c5烷基醇或任选经取代的c6~c
20
碳环芳基;r3、r5和r7各自独立地为任选经取代的c1~c20烷基、任选经取代的c1~c20杂烷基、任选经取代的c6~c20环烷基、任选经取代的c6~c
20
碳环芳基或任选经取代的c6~c
20
杂芳烷基,优选各自独立地为任选经取代的c1~c5烷基、任选经取代的c6~c20环烷基或任选经取代的c6~c
20
碳环芳基。
[0049]
步骤s1中,所述三(1,3-氧硫杂环戊烷-2-亚硫酰-5-甲基)异氰脲酸酯与1,8-二氨基-3,6-二氧杂辛烷的摩尔比优选为1:(2~2.5),如1:2、1:2.1、1:2.2、1:2.3、1:2.4、1:
2.5。所述开环缩聚反应的条件优选包括温度为室温,时间为20~30h,如20h、22h、25h、28h、30h。此外,所述开环缩聚反应在有机溶剂(如二甲基甲酰胺等)的存在下进行。
[0050]
步骤s2中,所述三(1,3-氧硫杂环戊烷-2-亚硫酰-5-甲基)异氰脲酸酯与烷氧基三聚氰胺的摩尔比优选为1:(0.8~1.2),如1:0.8、1:0.9、1:1.0、1:1.1、1:1.2。所述消去反应的条件优选包括温度为40~50℃,如40℃、42℃、45℃、48℃、50℃;时间为1~5h,如1h、2h、3h、4h、5h。
[0051]
步骤s2中,所述烷氧基三聚氰胺可以为高度醚化型烷氧基三聚氰胺,也可以为部分醚化型烷氧基三聚氰胺,具体选择如下:
[0052]
高度醚化型:
[0053][0054]
部分醚化型:
[0055]
[0056][0057]
所述可自交联巯基三聚氰胺聚合物的重均分子量优选为5000~15000,更优选为6000~14000,最优选为7000~10000,如7000、7500、8000、8500、9000、9500、10000。
[0058]
在本发明中,所述酸产生剂主要起做的作用是促进可自交联异氰脲酸酯聚合物的交联反应。所述酸产生剂可以使用常规的光酸发生剂(pag)和/或热酸发生剂(thermal acid generator),具体可以选自基于锍盐的化合物、基于碘盐的化合物、基于盐的化合物、有机磺酸等中的至少一种。所述酸产生剂的具体实例包括但不限于:十二烷基苯磺酸、对甲苯磺酸、苯二甲酰亚氨基三氟甲磺酸酯、苯二甲酰亚氨基三氟甲磺酸盐、二硝基苄基甲苯磺酸酯、二硝基苄基甲苯磺酸盐、正癸基二砜、萘基亚氨基三氟甲磺酸酯、萘基亚氨基三氟甲磺酸盐、二苯基碘三氟甲磺酸盐、二苯基碘全氟丁基磺酸盐、二苯基碘六氟磷酸盐、二苯基碘六氟砷酸盐、二苯基碘六氟锑酸盐、二苯基对甲氧基苯基锍三氟甲磺酸盐、二苯基对甲苯锍三氟甲磺酸盐、二苯基对叔丁基苯基锍三氟甲磺酸盐、二苯基对异丁基苯基锍三氟甲磺酸盐、三苯基锍三氟甲磺酸盐、三(对叔丁基苯基)锍三氟甲磺酸盐、二苯基对甲氧基苯基锍全氟丁基磺酸盐、二苯基对甲苯基锍全氟丁基磺酸盐、二苯基对叔丁基苯基锍全氟丁基磺酸盐、二苯基对异丁基苯基锍全氟丁基磺酸盐、三苯基锍全氟丁基磺酸盐、三对叔丁基苯基锍全氟丁基磺酸盐、六氟砷酸酯、六氟砷酸盐、三苯基锍六氟锑酸盐和二丁基萘基锍三氟甲磺酸盐中的至少一种。所述酸产生剂的用量可以占组合物总重量的0.01%~1%,优选占组合物总重量的0.01%~0.3%,更优选占组合物总重量的0.02%~0.2%,最优选占组合物总重量的0.03%~0.15%。
[0059]
所述有机溶剂可使用用于形成抗反射涂层组合物的常规有机溶剂,其具体实例包括但不限于:2-羟基异丁酸甲酯(hbm)、环己酮、环戊酮、丁内酯、二甲基乙酰胺、二甲基甲酰胺、二甲基亚砜、n-甲基吡咯烷酮(nmp)、四氢糠醇、丙二醇单甲醚(pgme)、丙二醇单甲醚乙酸酯(pgmea)、乳酸乙酯中的至少一种,优选选自2-羟基异丁酸甲酯、环戊酮、丙二醇单甲醚和丙二醇单甲醚乙酸酯中的至少一种。此外,所述有机溶剂的用量可以占组合物总重量的90%~99%,优选占组合物总重量的93%~98.7%,最优选占组合物总重量的95%~98.5%。
[0060]
所述表面活性剂可以为氟化表面活性剂和/或非氟化表面活性剂,优选为非离子型氟化表面活性剂。其中,所述非离子型氟化表面活性剂可以为全氟c4表面活性剂(如来自3m corporation的fc-4430和fc-4432表面活性剂)、氟二醇(如来自omnova的polyfox pf-636、pf-6320、pf-656和pf-6520氟表面活性剂)。所述表面活性剂的用量可以占组合物总重量的0~1%,优选占组合物总重量的0.001%~0.02%,更优选占组合物总重量的0.0015%~0.015%,最优选占组合物总重量的0.002%~0.01%。
[0061]
本发明提供的抗反射涂层组合物的制备方法包括将可自交联巯基三聚氰胺聚合物、酸产生剂、有机溶剂以及任选的表面活性剂混合均匀。其中,所述混合均匀的方法和条件可以为本领域的常规选择,对此本领域技术均能知悉,在此不作赘述。
[0062]
本发明提供的图案形成方法包括:将抗反射涂层组合物涂敷至基底上并进行热固化,以在基底上形成抗反射涂层;在抗反射涂层上形成光致抗蚀剂层;将所述光致抗蚀剂层曝光并显影以形成光刻胶图案。
[0063]
所述基底可以采用硅、二氧化硅或铝-氧化铝微电子晶片,也可以采用砷化镓、碳化硅、陶瓷、石英或铜基底,还适当地采用用于液晶显示器或其它平板显示器应用的基底,例如玻璃基底、氧化铟锡涂布基底等,还可采用用于光学和光电装置(例如波导)的基底。
[0064]
本发明中,将光致抗蚀剂组合物涂覆于抗反射涂层组合物上方之前,需要先将抗反射涂层组合物固化形成抗反射涂层。其中,所述固化的条件随着抗反射涂层组合物的组分而变化,确切地说,所述固化的条件取决于抗反射涂层组合物中采用的酸产生剂。典型的固化温度为90~240℃,优选为150~210℃。所述固化的条件优选地使得抗反射涂层组合物大体上不溶于使用的光致抗蚀剂溶剂以及显影剂溶液。
[0065]
以下将通过实施例对本发明进行详细描述。
[0066]
以下制备例中,三聚氰胺单元在聚合物中的摩尔百分比按照以下测试并计算得到:取0.5g制好的样品测c-nmr,之后将所得核磁谱图进行处理,具体地,对指定化学位移的峰进行积分,三聚氰胺单元化学位移179.2ppm,异氰尿酸酯单元化学位移150.7ppm,1,8-二氨基-3,6-二氧杂辛烷单元化学位移70.1ppm,三聚氰胺单元摩尔百分数=a/3/(a/3+b/3+c/2)
×
100%,其中,a表示化学位移179.2ppm处的积分数值,b表示化学位移150.7ppm处的积分数值,c表示化学位移70.1ppm处的积分数值。
[0067]
制备例1:制备预聚物s1
[0068]
在250ml圆底烧瓶中加入15g(28.5mmol)三(1,3-氧硫杂环戊烷-2-亚硫酰-5-甲基)异氰脲酸酯、10.5g(71.25mmol)1,8-二氨基-3,6-二氧杂辛烷和167.73g的二甲基甲酰胺(dmf),在室温下搅拌反应24h,之后将聚合物溶液按照体积比1:10的量滴加到大量的异丙醇和正庚烷混合溶液中(异丙醇和正庚烷体积比7:3),过滤得到固体聚合物,40℃真空干燥3h,得到预聚物s1,其具有式(4)所示的结构,重均分子量mw为4052,分散系数pdi为1.75。
[0069][0070]
式(4)中r1与式(1)中的r1定义相同。
[0071]
制备例2:制备可自交联巯基三聚氰胺聚合物j1
[0072]
往制备例1所得预聚物s140g中加入2-羟基异丁酸甲酯(hbm)60g搅拌至完全溶解得到预聚物溶液,再将该预聚物溶液在通氮气情况下加入到500ml三口烧瓶中,然后加热到45℃,之后加入9.8g(25.1mmol)六甲氧基甲基三聚氰胺(用hbm溶解成固含量为20%的溶液后使用)和0.48g(2.5mmol)ptsa(对甲苯磺酸),45℃搅拌下反应2h,接着往反应溶液中加入0.28g(2.75mmol)三乙胺,然后再加入180g的hbm溶剂稀释,得到聚合物溶液。
[0073]
将上述聚合物溶液按体积比1:10的量滴加到异丙醇和正庚烷混合溶液中(异丙醇和正庚烷体积比7:3),过滤得到固体聚合物,然后将固体聚合物按20%固含量溶解在hbm中,接着按体积比1:5的量滴加到异丙醇和正庚烷混合溶液中(异丙醇和正庚烷体积比7:3),搅拌30min,过滤得到固体聚合物,然后将固体聚合物在50℃真空干燥3h,得到聚合物白色固体粉末,即为可自交联巯基三聚氰胺聚合物j1,经核磁和红外检测,其结构如式(6)所示。该可自交联巯基三聚氰胺聚合物j1的重均分子量为8207,多分散性(pd)为1.88,按聚合物摩尔百分比三聚氰胺单元占比为11.5%。
[0074][0075]
制备例3:制备可自交联巯基三聚氰胺聚合物j2
[0076]
往制备例1所得预聚物s140g中加入2-羟基异丁酸甲酯(hbm)60g搅拌至完全溶解得到预聚物溶液,再将该预聚物溶液在通氮气情况下加入到500ml三口烧瓶中,然后加热到45℃,之后加入14.0g(25.1mmol)六异丙氧基甲基三聚氰胺(用hbm溶解成固含量为20%的溶液后使用)和0.48g(2.5mmol)ptsa(对甲苯磺酸),45℃搅拌下反应2h,接着往反应溶液中加入0.28g(2.75mmol)三乙胺,然后再加入180g的hbm溶剂稀释,得到聚合物溶液。
[0077]
将上述聚合物溶液按体积比1:10的量滴加到异丙醇和正庚烷混合溶液中(异丙醇和正庚烷体积比7:3),过滤得到固体聚合物,然后将固体聚合物按20%固含量溶解在hbm中,接着按体积比1:5的量滴加到异丙醇和正庚烷混合溶液中(异丙醇和正庚烷体积比7:3),搅拌30min,过滤得到固体聚合物,然后将固体聚合物在50℃真空干燥3h,得到聚合物白色固体粉末,即为可自交联巯基三聚氰胺聚合物j2,经核磁和红外检测,其结构如式(7)所示。该可自交联巯基三聚氰胺聚合物j2的重均分子量为8620,多分散性(pd)为1.92,按聚合物摩尔百分比三聚氰胺单元占比为10.6%。
[0078][0079]
制备例4:制备可自交联巯基三聚氰胺聚合物j3
[0080]
往制备例1所得预聚物s140g中加入2-羟基异丁酸甲酯(hbm)60g搅拌至完全溶解得到预聚物溶液,再将该预聚物溶液在通氮气情况下加入到500ml三口烧瓶中,然后加热到45℃,之后加入15.6g(25.1mmol)六丁氧基甲基三聚氰胺(用hbm溶解成固含量为20%的溶液后使用)和0.48g(2.5mmol)ptsa(对甲苯磺酸),45℃搅拌下反应2h,接着往反应溶液中加入0.28g(2.75mmol)三乙胺,然后再加入180g的hbm溶剂稀释,得到聚合物溶液。
[0081]
将上述聚合物溶液按体积比1:10的量滴加到异丙醇和正庚烷混合溶液中(异丙醇和正庚烷体积比7:3),过滤得到固体聚合物,然后将固体聚合物按20%固含量溶解在hbm中,接着按体积比1:5的量滴加到异丙醇和正庚烷混合溶液中(异丙醇和正庚烷体积比7:3),搅拌30min,过滤得到固体聚合物,然后将固体聚合物在50℃真空干燥3h,得到聚合物白色固体粉末,即为可自交联巯基三聚氰胺聚合物j3,经核磁和红外检测,其结构如式(8)所示。该可自交联巯基三聚氰胺聚合物j3的重均分子量为9155,多分散性(pd)为2.06,按聚合物摩尔百分比三聚氰胺单元占比为10.2%。
[0082][0083]
制备例5:制备可自交联巯基三聚氰胺聚合物j4
[0084]
往制备例1所得预聚物s140g中加入2-羟基异丁酸甲酯(hbm)60g搅拌至完全溶解得到预聚物溶液,再将该预聚物溶液在通氮气情况下加入到500ml三口烧瓶中,然后加热到45℃,之后加入19.7g(25.1mmol)六环己氧基甲基三聚氰胺(用hbm溶解成固含量为20%的
溶液后使用)和0.48g(2.5mmol)ptsa(对甲苯磺酸),45℃搅拌下反应2h,接着往反应溶液中加入0.28g(2.75mmol)三乙胺,然后再加入180g的hbm溶剂稀释,得到聚合物溶液。
[0085]
将上述聚合物溶液按体积比1:10的量滴加到异丙醇和正庚烷混合溶液中(异丙醇和正庚烷体积比7:3),过滤得到固体聚合物,然后将固体聚合物按20%固含量溶解在hbm中,接着按体积比1:5的量滴加到异丙醇和正庚烷混合溶液中(异丙醇和正庚烷体积比7:3),搅拌30min,过滤得到固体聚合物,然后将固体聚合物在50℃真空干燥3h,得到聚合物白色固体粉末,即为可自交联巯基三聚氰胺聚合物j4,经核磁和红外检测,其结构如式(9)所示。该可自交联巯基三聚氰胺聚合物j4的重均分子量为9500,多分散性(pd)为2.26,按聚合物摩尔百分比三聚氰胺单元占比为9.8%。
[0086][0087]
制备例6:制备可自交联巯基三聚氰胺聚合物j5
[0088]
往制备例1所得预聚物s140g中加入2-羟基异丁酸甲酯(hbm)60g搅拌至完全溶解得到预聚物溶液,再将该预聚物溶液在通氮气情况下加入到500ml三口烧瓶中,然后加热到45℃,之后加入9.4g(25.1mmol)六环己氧基甲基三聚氰胺(用hbm溶解成固含量为20%的溶液后使用)和0.48g(2.5mmol)ptsa(对甲苯磺酸),45℃搅拌下反应2h,接着往反应溶液中加入0.28g(2.75mmol)三乙胺,然后再加入180g的hbm溶剂稀释,得到聚合物溶液。
[0089]
将上述聚合物溶液按体积比1:10的量滴加到异丙醇和正庚烷混合溶液中(异丙醇和正庚烷体积比7:3),过滤得到固体聚合物,然后将固体聚合物按20%固含量溶解在hbm中,接着按体积比1:5的量滴加到异丙醇和正庚烷混合溶液中(异丙醇和正庚烷体积比7:3),搅拌30min,过滤得到固体聚合物,然后将固体聚合物在50℃真空干燥3h,得到聚合物白色固体粉末,即为可自交联巯基三聚氰胺聚合物j5,经核磁和红外检测,其结构如式(10)所示。该可自交联巯基三聚氰胺聚合物j5的重均分子量为7860,多分散性(pd)为1.80,按聚合物摩尔百分比三聚氰胺单元占比为11.5%。
[0090][0091]
制备例7:制备可自交联巯基三聚氰胺聚合物j6
[0092]
往制备例1所得预聚物s140g中加入2-羟基异丁酸甲酯(hbm)60g搅拌至完全溶解得到预聚物溶液,再将该预聚物溶液在通氮气情况下加入到500ml三口烧瓶中,然后加热到45℃,之后加入11.9g(25.1mmol)六环己氧基甲基三聚氰胺(用hbm溶解成固含量为20%的溶液后使用)和0.48g(2.5mmol)ptsa(对甲苯磺酸),45℃搅拌下反应2h,接着往反应溶液中加入0.28g(2.75mmol)三乙胺,然后再加入180g的hbm溶剂稀释,得到聚合物溶液。
[0093]
将上述聚合物溶液按体积比1:10的量滴加到异丙醇和正庚烷混合溶液中(异丙醇和正庚烷体积比7:3),过滤得到固体聚合物,然后将固体聚合物按20%固含量溶解在hbm中,接着按体积比1:5的量滴加到异丙醇和正庚烷混合溶液中(异丙醇和正庚烷体积比7:3),搅拌30min,过滤得到固体聚合物,然后将固体聚合物在50℃真空干燥3h,得到聚合物白色固体粉末,即为可自交联巯基三聚氰胺聚合物j6,经核磁和红外检测,其结构如式(11)所示。该可自交联巯基三聚氰胺聚合物j6的重均分子量为8006,多分散性(pd)为1.81,按聚合物摩尔百分比三聚氰胺单元占比为10.8%。
[0094][0095]
对比制备例1:制备参比异氰尿酸酯聚合物dj1
[0096]
往制备例1所得预聚物s140g溶于四氢呋喃中,加入三乙胺,之后将反应温度降低至0℃后滴加过量的苯甲酰氯,滴加完成后将温度升至室温反应20小时,得到参比异氰尿酸酯聚合物dj1,其重均分子量mw为5103,多分散性pd为1.89。
[0097]
对比制备例2:制备参比异氰尿酸酯聚合物dj2
[0098]
按照制备例1的方法制备可自交联巯基三聚氰胺聚合物,不同的是,将六甲氧基甲基三聚氰胺采用相同摩尔量的四甲氧基甲基甘脲替代,其余条件与制备例2相同,得到参比可自交联巯基甘脲聚合物dj2。
[0099]
实施例1:制备抗反射涂层组合物z1
[0100]
将2.95g由制备例2所得聚合物j1、0.03g p-tsa苄基铵盐、0.003g来自3m的氟化物表面活性剂fc-4430和97.00g 2-羟基异丁酸甲酯(hbm)混合均匀,之后经由具有0.45微米孔径的ptfe微过滤器过滤,获得抗反射涂层组合物z1。
[0101]
实施例2:制备抗反射涂层组合物z2
[0102]
将2.95g由制备例3所得聚合物j2、0.03g p-tsa苄基铵盐、0.003g来自3m的氟化物表面活性剂fc-4430和97.00g 2-羟基异丁酸甲酯(hbm)混合均匀,之后经由具有0.45微米孔径的ptfe微过滤器过滤,获得抗反射涂层组合物z2。
[0103]
实施例3:制备抗反射涂层组合物z3
[0104]
将2.95g由制备例4所得聚合物j3、0.03g p-tsa苄基铵盐、0.003g来自3m的氟化物表面活性剂fc-4430和97.00g 2-羟基异丁酸甲酯(hbm)混合均匀,之后经由具有0.45微米孔径的ptfe微过滤器过滤,获得抗反射涂层组合物z3。
[0105]
实施例4:制备抗反射涂层组合物z4
[0106]
将2.95g由制备例5所得聚合物j4、0.03g p-tsa苄基铵盐、0.003g来自3m的氟化物表面活性剂fc-4430和97.00g 2-羟基异丁酸甲酯(hbm)混合均匀,之后经由具有0.45微米孔径的ptfe微过滤器过滤,获得抗反射涂层组合物z4。
[0107]
实施例5:制备抗反射涂层组合物z5
[0108]
将2.95g由制备例6所得聚合物j5、0.03g p-tsa苄基铵盐、0.003g来自3m的氟化物表面活性剂fc-4430和97.00g 2-羟基异丁酸甲酯(hbm)混合均匀,之后经由具有0.45微米孔径的ptfe微过滤器过滤,获得抗反射涂层组合物z5。
[0109]
实施例6:制备抗反射涂层组合物z6
[0110]
将2.95g由制备例7所得聚合物j6、0.03g p-tsa苄基铵盐、0.003g来自3m的氟化物表面活性剂fc-4430和97.00g 2-羟基异丁酸甲酯(hbm)混合均匀,之后经由具有0.45微米孔径的ptfe微过滤器过滤,获得抗反射涂层组合物z6。
[0111]
对比例1:制备参比抗反射涂层组合物dz1
[0112]
将2.65g由制备例1所得聚合物s1、0.3g六甲氧基甲基三聚氰胺、0.03gp-tsa苄基铵盐、0.003g来自3m的氟化物表面活性剂fc-4430和97.00g 2-羟基异丁酸甲酯(hbm)混合均匀,之后经由具有0.45微米孔径的ptfe微过滤器过滤,获得参比抗反射涂层组合物d1。
[0113]
对比例2:制备抗反射涂层组合物dz2
[0114]
将2.65g由对比制备例1所得参比异氰尿酸酯聚合物dj1、0.3g六甲氧基甲基三聚氰胺、0.03g p-tsa苄基铵盐、0.003g来自3m的氟化物表面活性剂fc-4430和97.00g 2-羟基异丁酸甲酯(hbm)混合均匀,之后经由具有0.45微米孔径的ptfe微过滤器过滤,获得参比抗反射涂层组合物d2。
[0115]
对比例3:制备抗反射涂层组合物dz3
[0116]
按照实施例1的方法制备抗反射涂层组合物,不同的是,将由制备例2所得聚合物j1采用相同摩尔量的由对比制备例2所得参比可自交联巯基甘脲聚合物dj2替代,其余条件
与实施例1相同,得到参比抗反射涂层组合物dz3。
[0117]
测试例
[0118]
将以上实施例和对比例所得抗反射涂层组合物旋涂至硅片的蚀刻层上,之后在200℃下烘烤60秒以形成厚度为的抗反射涂层,再使用椭率计(制造商:j.a.woolam,名称:vuv-303)检测该抗反射涂层的折射率n、消光系数k和刻蚀速率。所得结果见表1。
[0119]
表1
[0120]
序号折射率n消光系数k刻蚀速率nm/s实施例11.960.4114.5实施例21.970.4013.6实施例31.970.4113.8实施例41.960.4012.8实施例51.970.4214.2实施例61.970.4114.0对比例11.960.429.5对比例21.970.419.8对比例31.960.419.1
[0121]
从表1的结果可以看出,本发明提供的抗反射涂层组合物烘烤固化形成的涂层兼具有高折射率、消光系数和优异的刻蚀速率。
[0122]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在不脱离本发明的原理和宗旨的情况下在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。