1.本发明属于发光材料领域,具体地,涉及发光性能稳定的量子点复合转光材料及制备发光性能稳定的量子点复合转光材料的磁控溅射法。
背景技术:
2.植物的光合作用是在叶绿体中进行的,对光的要求除了光强度外,光谱组成也是很重要的因素。叶绿素主要吸收太阳光中的蓝紫光(波长范围为400~480nm)和红橙光(波长范围为600~680nm),黄绿光(波长范围为510~580nm)大多被反射而不被吸收。农用转光材料能吸收日光中的紫外光(波长范围为200~400nm)或黄绿光,发射蓝紫光或红橙光,提高植物光合作用效率。
3.量子点作为一种独特的新型发光材料,因其光色纯正、发光颜色可调控、量子产率高、物化性质稳定、良好的生物相容性等优点而受到广泛关注。将量子点应用于农用转光材料中,有利于提高作物的光能利用率,促进植物的光合作用。在量子点材料的相关研究中,人们通常用对量子点进行表面修饰,主要使用化学气相沉积法和物理蒸汽沉积法,在其表面形成一层包覆层,这种做法虽然能够提高量子点的发光效率,使得量子点性质更加稳定,然而具有一层包覆层的量子点的发光效率的提高是在有限的范围内,发光效率很容易就达到极值不能再进一步提高。而且采用化学气相沉积法和物理蒸汽沉积法所形成的包膜的均匀性和致密性都不足,难以对量子点起到长效的保护作用。将其暴露在空气中仍会和空气中的水、氧气以及二氧化碳发生反应导致发光性能下降,产品的使用寿命较短。因此需要对量子点发光材料进行进一步研究,以进一步增强其发光效率的同时提高其耐候性。
4.溅射法是利用直流或者高频电场使惰性气体(通常为ar气)电离,电离产生的正离子在电场作用下轰击阴极靶材通过能量交换将能量传递给靶材原子,被溅射出来的原子以一定的动能射向衬底并在衬底上沉积形成薄膜的方法。磁控溅射技术是在普通溅射技术的基础上,为了提高成膜速率和降低工作气压,在阴极靶材背面放置了永久磁铁,从而形成磁场的方法。溅射室抽到一定的真空后,通入0.1~10pa的惰性气体(通常为ar气)作为溅射气体。在阴极靶材和阳极衬底之间加上一个正交的磁场和电场,电场和磁场方向相互垂直。在电场作用下ar原子发生电离,产生等离子体辉光放电。被电离的ar
+
在电场作用下加速飞向溅射靶材,并轰击靶材表面;二次电子则在正交的电磁场作用下以摆线和螺旋线的复合方式沿靶表面做圆周运动,其运动路径得到延长,从而增加了同ar气的碰撞次数,提高了电子的电离效率。ms技术获取的薄膜具备以下特点:沉积速率高,沉积温度低,对膜层的损伤小,工作气压较低,沉积制备的薄膜均匀致密、表面光洁度高、纯度高和黏附性能强的特点,适用于高效、大批的工业化生产中。目前,磁控溅射技术广泛应用于平板显示器件、太阳能电池、微波与射频屏蔽装置与器件、传感器等,然而,将磁控溅射技术在发光材料领域中的应用仍存在较大的空白。
技术实现要素:
5.根据本发明的第一个方面,提供
6.一种发光性能稳定的量子点复合转光材料,包括:基底,基底为内部填充有量子点的聚硅氧烷微球;磁控溅射膜,磁控溅射膜为通过磁控溅射法生成成膜物质并使成膜物质沉积在基底的表面而形成;成膜物质通过金属靶材和反应气体反应生成,靶材选自单晶硅、金属钛、金属铝或上述物料中的任意组合而形成的合金,反应气体选自o2、n2中的至少一种。
7.优选地,磁控溅射膜的厚度为100~2000nm。
8.优选地,磁控溅射膜由sio2、tio2、tin、ticn、al2o3中的至少一种物质所构成。
9.本发明采用磁控溅射法对量子点复合转光材料进行表面包覆,通过靶材溅射出的靶原子与反应气体反应形成成膜物质,成膜物质沉积到基底的表面形成致密、均一的磁控溅射膜。基于磁控溅射法的成膜特性,原料选择范围广,只要能制成靶材的原材料,均能发生溅射生成靶原子,并且可以选自包含多种原子的合金材料,因此能可控地在基底表面形成不同成分和不同性质的磁控溅射膜且可以通过调控工艺参数制得厚度较薄的磁控溅射膜。采用本发明采用单晶硅、金属钛、金属铝或上述物料中的任意组合作为靶材,以o2、n2作为反应气体,可以生成sio2、tio2、tin、ticn、al2o3成膜物质中的至少一种,进而可以形成由两种以上上述成膜物质的复合沉积构成的磁控溅射膜。上述成膜物质化学性质稳定,能够将基底与空气隔绝,避免其与空气中的物质发生反应。由于上述磁控溅射膜表面粒子的尺寸在纳米级别,具有良好的光学性能,将其用于包覆量子点复合转光材料基底,在起到增强基底耐候性的同时又基本不会影响其原本的的发光性能。
10.优选地,磁控溅射膜由sio2和al2o3共同构成,在磁控溅射膜中,硅元素和铝元素的摩尔比介于3.5~11。由此形成的磁控溅射膜中含有大量的sio2,能够降低磁控溅射膜与聚硅氧烷微球基底的界面差异,有利于磁控溅射膜与基底表面的复合,而且sio2具有良好的透光性,而通过在sio2中掺杂al2o3有利于形成致密、强韧的保护膜层,而通过在al2o3中掺杂sio2,能够降低磁控溅射膜与基底表面的界面差异,有利于磁控溅射膜与基底表面的复合。通过磁控溅射法能够准确地硅元素和铝元素的摩尔比控制在上述范围内,由此得到的磁控溅射膜既能够对量子点复合转光材料起到优异的保护效果,有效地延长了量子点复合转光材料的使用寿命,又能够保有量子点发光材料本身优越的发光特性。
11.优选地,上述发光性能稳定的量子点复合转光材料,在200~400nm、480~600nm光谱范围内有至少一个吸收峰。
12.优选地,上述发光性能稳定的量子点复合转光材料在600~700nm的光谱范围内有至少一个发射峰。
13.植物进行光合作用所吸收的波长范围蓝紫光(波长范围为400~480nm)和红橙光(波长范围为600~680nm),本发明提供的量子点复合转光材料的吸收光谱在波长范围200~400nm和480~600nm至少有一个吸收峰,并且发射光谱在600~700nm的光谱范围内有至少一个发射峰。从而能将植物不能吸收利用的波段的光波转化成植物光合作用所需的波长的光波,能够有效促进植物的生长发育。
14.优选地,量子点为cuins2。
15.优选地,上述量子点的表面被zns修饰。
16.优选地,上述基底的粒径小于10nm。
17.本发明所使用的基底为内部填充有量子点的聚硅氧烷微球,具体通过以下方式制备:
18.1.制备cuins2@zns量子点,具体包括以下步骤:
19.(1)将铜源化合物、铟源化合物、第一硫源化合物和水混合放入反应釜中,搅拌下调节ph为10-12;再加入第二硫源化合物得到澄清的混合溶液,将上述澄清的混合溶液加热并保温至少10~24h后得到cuins2量子点水溶液;
20.(2)取降温后的cuins2量子点水溶液,向其中加入锌源化合物、第二硫源化合物,充分混合后,加入保温6~12h,离心后将得到的沉淀物重新溶于水中,得到cuins2@zns量子点溶液。
21.优选地,上述铜源化合物为二水氯化铜。
22.优选地,上述铟源化合物为四水合三氯化铟。
23.优选地,上述第一硫源化合物和第二硫源化合物分别为3-巯基丙酸和硫脲。
24.优选地,上述锌源化合物选自硫酸锌、醋酸锌、氯化锌的一种或多种。
25.优选地,上述步骤(1)使用氢氧化钠溶液调节ph为11。
26.优选地,上述铜源化合物、铟源化合物和第二硫源化合物的摩尔比为1:1:2。
27.2.制备基底psi@(cuins2@zns),具体包括以下步骤:
28.按重量计,将硅烷单体加入量子点溶液中,搅拌后静置得到白色沉淀物,将其抽滤和洗涤后烘干,得到psi@(cuins2@zns)。
29.优选地,上述硅烷单体选自含甲氧基或乙氧基的硅烷单体。
30.优选地,上述含甲氧基或乙氧基的硅烷单体选自脲丙基三甲氧基硅烷、脲丙基三乙氧基硅烷、甲基苯基二甲氧基硅烷、二甲氧基甲基乙烯基硅烷、正丙基三甲氧基硅烷、正十二烷基三甲氧基硅烷、三甲氧基(7-辛烯-1-基)硅烷中的任意一种。
31.优选地,按重量比计,上述硅烷单体:上述量子点=8~10:1。
32.本发明采用内部填充有cuins2量子点的聚硅氧烷微球作为基底,作为新型转光材料,cuins2量子点不含有毒元素,能够被黄绿光激发,发射出植物生长所需的红橙光。用zns对cuins2量子点进行表面修饰,能够进一步提高其发光效率。用聚硅氧烷包覆的cuins2@zns的耐候性和发光的浓度上限能够进一步提高。
33.根据本发明的另一个方面,提供一种制备发光性能稳定的量子点复合转光材料的磁控溅射法,其特征在于,包括以下步骤:步骤一,以惰性气体轰击靶材以使靶材溅射靶原子,形成辉光,其中,靶材为单晶硅、金属钛、金属铝或上述物料中的任意组合而形成的合金;步骤二,通入反应气体与靶原子发生反应形成成膜物质,成膜物质沉积到基底的表面形成包覆膜,其中,反应气体选自o2、n2,基底为内部填充有量子点的聚硅氧烷微球。
34.优选地,上述磁控溅射法还包括预备步骤:将基底置于真空度为10
-4
~10
-3
pa的反应区域内,加热基底至100~300℃。
35.优选地,靶材为单晶硅、金属铝或上述物料中的任意组合而形成的合金。
36.优选地,在步骤一中,惰性气体的流速为10~100sccm。
37.优选地,在步骤二中,将反应气体的流速控制在1~10sccm,溅射功率为50~200w。
38.可选地,溅射时间为1~4h。
附图说明
39.图1为以下实施例所采用的psi@(cuins2@zns)基底材料的荧光光谱图;
40.图2为测试例2中所涉及的荧光强度测试结果统计图。
具体实施方式
41.为了使本技术领域的人员更好地理解本发明方案,下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分的实施例,而不是全部的实施例。
42.以下实施例1~4和对比实施例1~5中所使用的基底psi@(cuins2@zns)通过以下方法制备:
43.制备cuins2量子点溶液:按照摩尔比n(in):n(cu):n(s)=1:1:12,称取四水三氯化铟0.0453g、二水氯化铜0.0263g、3-巯基丙酸0.1986g和14ml水混合后放入反应釜中,搅拌下滴入氢氧化钠溶液调节ph至10,将上述溶液转移至四氟乙烯反应釜中,置于烘箱130℃下加热并保温24h,即得到cuins2量子点溶液。
44.制备cuins2@zns:取上述cuins2量子点溶液,向其中加入硫酸锌、硫脲,充分混合后,于140℃下加热并保温12h,离心后将得到的沉淀物重新溶于水中,得到质量分数约10%的cuins2@zns量子点溶液。
45.制备psi@(cuins2@zns):取2g上述cuins2@zns量子点溶液,加入到含有1.2ml脲丙基三乙氧基硅烷的40ml水溶液中,磁力搅拌30min后静置4h,将产生白色沉淀物经过抽滤、洗涤、烘干,最后得到psi@(cuins2@zns)基底材料。所制得的psi@(cuins2@zns)基底材料的荧光光谱图如图1所示。
46.实施例1
47.本实施例以psi@(cuins2@zns)作为基底,通过磁控溅射法,采用单晶硅作为靶材,按照以下方法制备表面包覆有sio2磁控溅射膜的psi@(cuins2@zns)量子点复合转光材料:
48.(1)抽真空:将psi@(cuins2@zns)粉末放入磁控溅射镀膜真空室的振动样品台上,对真空室进行抽真空至10-3
pa;
49.(2)加热基体:将psi@(cuins2@zns)粉末加热至150℃;
50.(3)通入溅射气体氩气:等到温度稳定后,先向真空室内通入氩气,设置氩气的流速为50sccm,并调节真空室内气压为6.5pa;
51.(4)预溅射:打开中频电源,先用小功率进行预溅射,以去除靶材表面的杂质;
52.(5)溅射镀膜:等到辉光稳定后,通入o2,流量为5sccm,并调大溅射功率为60w,打开靶材上的挡板,进行溅射1h,制备500nm的sio2磁控溅射膜。
53.本实施例以上述制得的表面包覆有sio2磁控溅射层的psi@(cuins2@zns)量子点复合转光材料作为参试量子点复合发光材料。
54.实施例2
55.本实施例以psi@(cuins2@zns)作为基底,通过磁控溅射法,采用金属铝作为靶材,按照以下方法制备表面包覆有al2o3磁控溅射膜的psi@(cuins2@zns)量子点复合转光材料:
56.(1)抽真空:将psi@(cuins2@zns)
+
粉末放入磁控溅射镀膜真空室的振动样品台上,对真空室进行抽真空至5
×
10-4
pa;
57.(2)加热基体:将硫化物粉末加热至180℃;
58.(3)通入溅射气体氩气:等到温度稳定后,先向真空室内通入氩气,设置氩气的流速为55sccm,并调节真空室内气压为7.5pa;
59.(4)预溅射:打开中频电源,先用小功率进行预溅射,以去除靶材表面的杂质;
60.(5)溅射镀膜:等到辉光稳定后,通入o2,流量为10sccm,并调大溅射功率为110w,打开靶材上的挡板,进行溅射2.5h,制备500nm的al2o3磁控溅射膜。
61.本实施例以上述制得的表面包覆有al2o3磁控溅射层的psi@(cuins2@zns)量子点复合转光材料作为参试量子点复合发光材料。
62.实施例3
63.本实施例以psi@(cuins2@zns)作为基底,通过磁控溅射法,采用铝硅合金作为靶材,在本实施例中所采用的靶材中依次包括了铝含量为约为25wt%、20wt%、18wt%和15wt%的铝硅合金,按照摩尔比换算,上述靶材中,硅元素和铝元素的摩尔比依次对应为2.89、3.85、4.39、5.46,分别采用上述铝含量不同的铝硅合金靶材,按照以下方法制备表面包覆有sio
2-al2o3磁控溅射膜的psi@(cuins2@zns)量子点复合转光材料:
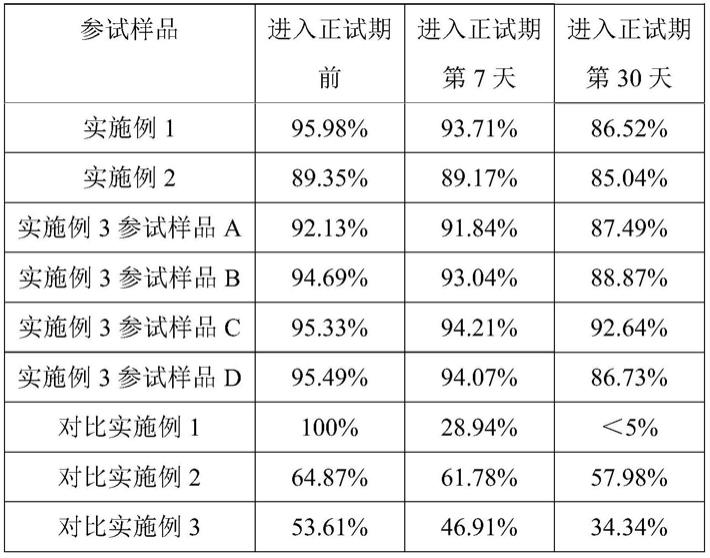
64.(1)抽真空:将psi@(cuins2@zns)粉末放入磁控溅射镀膜真空室的振动样品台上,对真空室进行抽真空至10-4
pa;
65.(2)加热基体:将psi@(cuins2@zns)粉末加热至200℃;
66.(3)通入溅射气体氩气:等到温度稳定后,先向真空室内通入氩气,设置氩气的流速为45sccm,并调节真空室内气压为4pa;
67.(4)预溅射:打开中频电源,先用小功率进行预溅射,以去除靶材表面的杂质;
68.(5)溅射镀膜:等到辉光稳定后,通入o2,流量为7sccm,并调大溅射功率为75w,打开靶材上的挡板,进行溅射3h,制备500nm的sio
2-al2o3磁控溅射膜。
69.本实施例以上述制得的表面包覆有sio
2-al2o3磁控溅射层的psi@(cuins2@zns)量子点复合转光材料作为参试量子点复合发光材料。根据所采用的靶材不同,将本实施例所制得的参试量子点复合发光材料进行标号,参试量子点复合发光材料与其所采用的靶材的对应关系为:采用铝含量为约为25wt%的铝硅合金靶材制得的参试量子点复合发光材料,标记为参试样品a;采用铝含量为约为20wt%的铝硅合金靶材制得的参试量子点复合发光材料,标记为参试样品b;采用铝含量为约为18wt%的铝硅合金靶材制得的参试量子点复合发光材料,标记为参试样品c;采用铝含量为约为15wt%的铝硅合金靶材制得的参试量子点复合发光材料,标记为参试样品d。
70.对比实施例1
71.该对比实施例以未经包覆处理的psi@(cuins2@zns)粉末作为参试量子点复合发光材料。
72.对比实施例2
73.利用溶胶凝胶法制备具有sio2微球结构的量子点复合转光材料:烧杯中加入2.5ml乙醇和4ml去离子水,以每分钟0.6ml的速率滴入含有5%的正硅酸乙酯的水溶液3ml,往由此形成的水溶液中加入2g上述cuins2@zns量子点溶液,在60℃下搅拌30min后,逐滴滴加少量氨水,调节ph为9-10,继续搅拌45min,然后在80℃下真空干燥2h,并在马弗炉中500℃煅烧1h,制得具有sio2微球结构的量子点复合转光材料sio2@(cuins2@zns),(cuins2@
zns)填充在sio2微球结构之中。
74.以上述sio2@(cuins2@zns)替换实施例1中的psi@(cuins2@zns)作为基底,沿用实施例1所提供的磁控溅射工艺制备表面包覆有sio2磁控溅射层的psi@(cuins2@zns)量子点复合转光材料。
75.本实施例以上述制得的表面包覆有sio2磁控溅射层的psi@(cuins2@zns)量子点复合转光材料作为参试量子点复合发光材料。
76.对比实施例3
77.采用溶胶凝胶法制备表面包覆sio
2-al2o3包覆层的量子点复合转光材料psi@(cuins2@zns):烧杯中加入2.5ml乙醇和4ml去离子水,同时向由此形成的乙醇溶液中滴加含有5%的正硅酸乙酯的水溶液和0.1m硝酸铝溶液,正硅酸乙酯和硝酸铝的用量比满足硅元素和铝元素的摩尔比为si:al=2:16.5,往由此形成的水溶液中加入psi@(cuins2@zns)粉末(7nm),常温下搅拌30min,逐滴滴加少量氨水,调节ph为9-10,继续搅拌45min,然后在80℃下真空干燥2h,并在马弗炉中1000℃煅烧2h,得到表面包覆sio
2-al2o3包覆层的量子点复合转光材料psi@(cuins2@zns)。
78.本实施例以上述制得的表面包覆sio
2-al2o3包覆层的量子点复合转光材料psi@(cuins2@zns)作为参试量子点复合发光材料,该包覆层的厚度约为500nm。
79.测试例1
80.将实施例1~4制得的量子点复合转光材料粉体和对比实施例1~5制得的转光材料粉体进行耐湿劣化实验,具体的实验设置方式如下:
81.正试期,将参试粉体置于温度为25℃,相对湿度为80%的温箱中进行耐湿劣化实验测试。对进入正试期前和进入正试期的参试样品进行荧光光谱测试,在激发波长为540nm的测试条件下,测得参试粉体的发射光谱图,记录参试粉体的发射光谱在波长为640nm处的发光强度。
82.发光性能保持率为参试粉体在不同测试时间的发光强度与对比例实施例1参试粉体在进入正式期前的发光强度之比,具体测试结果如表1所示。
83.表1实施例1~4和对比实施例1~6量子点转光材料的发光性能保持率
[0084][0085]
具体测试结果显示,本发明实施例1~3运用磁控溅射法对基底进行表面包覆,能够制备得到的发光性能稳定的量子点复合转光材料。与对比实施例1提供的未经包覆的cuins2@zns相比,实施例1~3、对比实施例2和对比实施例3分分别采用不同的工艺在psi@(cuins2@zns)表面形成保护膜,会使cuins2@zns的发光强度发生一定量的折减。实施例1~3用于形成保护膜的方法为磁控溅射法,由于al2o3的透光性较sio2的透光性差,因此,实施例2所制得的参试量子点复合转光材料在进入正试期时的发光强度相对较低,而实施例1和实施例3在psi@(cuins2@zns)量子的表面形成了含有大量sio2的磁控溅射层,包覆前后,psi@(cuins2@zns)的发光性能几乎不受影响。在耐湿劣化实验中,与正试期开始时相比,在进入正试期后第7天时,实施例2和实施例3所提供的参试样品的发光性能基本不受影响,且经过30天的耐湿劣化实验处理之后,其发光强度仍能保持与初始发光强度相当。与采用纯sio2磁控溅射膜包覆的实施例1、采用纯al2o3磁控溅射膜包覆的实施例2相比,实施例3所制得的几种表面具有sio
2-al2o3磁控溅射膜的参试样品呈现出更加的发光性能稳定性。其中,实施例3所制得的参试样品b、参试样品c和参试样品d的初始发光强度基本上能够达到与实施例1的参试样品相当的水平,然而,通过al2o3的掺杂,随着进入正试期时间的增长,实施例3所制得的参试产品所展现出的发光性能稳定性明显更高,其中,psi@(cuins2@zns)基底表面所形成的磁控溅射层中的硅元素和铝元素的含量比会对基底的发光性能产生一定的影响,实施例3通过采用不同的铝硅合金靶材而实现了对所形成的磁控溅射层中的硅元素和铝元素的相对比例进行精确的调控,其中,采用铝元素含量为18wt%左右的铝硅合金靶材所制得的参试样品c所具备的放光性能稳定性最佳,在众多参试样品中,实施例3的参试产品c表现出明显更为优异的发光性能稳定性。与实施例1形成对比,对比实施例2将cuins2@zns填充在二氧化硅微球中,由此会使cuins2@zns的发光强度发生明显的下降。通过对比实施例3和对比实施例3,虽然上述两个实施例都的基底表面都被sio
2-al2o3层包覆,然而,对比实施例3采用溶胶凝胶法形成的sio
2-al2o3层包覆对基底的保护效果不佳,经过劣化实验处理后,对比实施例3的参试量子点复合转光材料的发光强度剧烈下降。因此通过溶胶凝胶法制
备得到的样品不具备稳定的发光性能。
[0086]
测试例2
[0087]
1.参试梯度溶液的配制
[0088]
称取一定质量的实施例1所采用的参试量子点复合发光材料(表面包覆有sio2磁控溅射层的psi@(cuins2@zns)量子点复合转光材料)溶于正己烷中,分别配成物质的量浓度为1μmol.l-1
、1.5μmol.l-1
、2μmol.l-1
、3μmol.l-1
、5μmol.l-1
的溶液。
[0089]
称取一定质量的对比实施例1所采用的参试量子点复合发光材料(未经包覆的cuins2@zns粉末)溶于正己烷中,分别配成物质的量浓度为1μmol.l-1
、1.5μmol.l-1
、2μmol.l-1
、3μmol.l-1
、5μmol.l-1
的溶液。
[0090]
2.荧光强度测试
[0091]
对本测试例的参试梯度溶液进行荧光光谱测试,在激发波长为540nm的测试条件下,测得参试样品的发射光谱图,记录参试样品的发射光谱在波长为640nm处的发光强度。以利用未经包覆的cuins2@zns粉末配制得到浓度为1μmol.l-1
的参试溶液作为基准溶液,以基准溶液的发光强度为100%,其他参试溶液的发光性能的计算方法为参试溶液与基准溶液的发光强度之比。荧光测试结果如图2所示,随着量子点复合材料的浓度增加,未经包覆的cuins2@zns所形成的参试溶液的发光强度呈现先增长后下降的趋势,在浓度为2μmol.l-1
达到发光强度的上限,然而,表面包覆有sio2磁控溅射层的psi@(cuins2@zns)量子点复合转光材料的发光强度一直呈现增长的趋势,结果表明,本发明通过对cuins2@zns进行包覆,能够增强量子点的分散性,进而增大了量子点复合转光材料发光强度的浓度上限,优化了量子点复合转光材料的发光效果。
[0092]
以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。