1.本发明属于有机合成技术领域,尤其涉及一种酰胺类化合物或其药学上可接受的盐及其制备方法和应用。
背景技术:
2.鼠类肉瘤病毒癌基因(ras)蛋白是gtp酶家族中的一个重要成员,包括nras,hras和kras。这些酶在细胞外信号转导,增殖,凋亡和分化中起重要作用。kras与鸟嘌呤三核苷酸磷酸(gtp)结合为活性构象,与鸟嘌呤二核苷酸磷酸(gdp)结合为无活性构象。突变的kras蛋白和gtp紧密结合使kras蛋白处于异常的持续激活构象,导致下游信号通路持续性激活。kras抑制剂通过识别突变kras,阻断kras/gef相互作用,抑制kras下游效应因子从而产生抗肿瘤活性。然而,由于体内高水平的gtp和kras与gtp强有力的结合作用力,导致靶向kras变得非常困难。此外,kras蛋白是一个狭长的平坦口袋,这导致了基于kras药物的设计变得困难。kras被称为“不可成药靶点”。
3.近年来,针对kras突变的研究主要集中在kras g12c,突变的半胱氨酸使得药物能够通过共价结合的方式进行设计。kras g12c抑制剂通过与半胱氨酸(cys)发生迈克尔加成,导致蛋白质构象改变,从而使krasg12c停留在无活性kras-gdp构象,从而抑制下游蛋白,包括ras-mek-erk信号通路,产生抗肿瘤作用。目前kras g12c抑制剂amg510、mrtx849等已经进入临床,有望成为下一个重磅药物。尽管kras g12c抑制剂发展迅速,然而还是出现了一些耐药性的问题,这主要是肿瘤细胞kras旁路信号的激活。
4.因此,有必要提供一种抗肿瘤效果更好的化合物。
技术实现要素:
5.本发明旨在至少解决现有技术中存在的上述技术问题之一。为此,本发明提供了一种酰胺类化合物或其药学上可接受的盐。
6.本发明还提供了所述酰胺类化合物或其药学上可接受的盐的制备方法。
7.本发明还提供了所述酰胺类化合物或其药学上可接受的盐的应用。
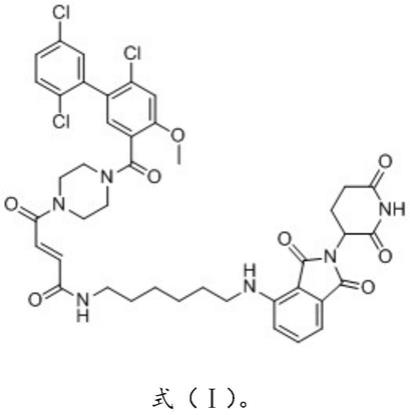
8.本发明的第一方面提供了一种酰胺类化合物或其药学上可接受的盐,所述酰胺类化合物的结构式如式(ⅰ)所示:
[0009][0010]
本发明关于酰胺类化合物或其药学上可接受的盐的技术方案,至少具有以下有益效果:
[0011]
本发明的酰胺类化合物能够结合e3泛素化连接酶,招募靶蛋白、引起目标蛋白的泛素化和降解,通过实现降解krasg12c蛋白,且能够特异性降解krasg12c,从而产生抗肿瘤活性,抗肿瘤活性效果好。
[0012]
本发明的第二方面提供了一种酰胺类化合物或其药学上可接受的盐的制备方法,包括如下步骤:
[0013]
s1.将化合物1、boc-己二胺和n,n-二异丙基乙胺加入到第一有机溶剂中进行反应得到化合物2,化合物2在酸性条件下得到化合物3;
[0014]
s2.在第二有机溶剂中加入化合物4和2,5-二氯苯硼酸,在钯催化剂和无机碱的条件下进行反应得到化合物5;化合物5在酸性条件下得到化合物6;
[0015]
s3.将化合物6、顺丁烯二酸酐和n,n-二异丙基乙胺在第三有机溶剂中反应得到化合物7;
[0016]
s4.将化合物7、化合物3、1-羟基苯并三唑和三乙胺在第四有机溶剂中反应得到式(ⅰ)化合物;
[0017]
所述化合物1~7的结构式如下:
[0018]
[0019]
根据本发明的一些实施方式,所述化合物1通过如下步骤制得:
[0020]
将3-氟磷酸酐和3-氨基-2,6-哌啶二酮加入到溶剂和有机碱中进行加热反应,即得化合物1。
[0021]
根据本发明的一些实施方式,所述化合物4通过如下步骤制得:
[0022]
将4-氯-5-碘-2-甲氧基苯甲酸、boc哌嗪和n,n-二异丙基乙胺加入到第五有机溶剂中进行反应,即得到化合物4。
[0023]
根据本发明的一些实施方式,所述第一有机溶剂、第二有机溶剂、第三有机溶剂、第四有机溶剂和第五有机溶剂分别独立地选自n,n-二甲基甲酰胺、甲苯或1,4二氧六环中至少一种。
[0024]
根据本发明的一些实施方式,步骤s2中的所述钯催化剂为四三苯基膦钯或[1,1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物中一种。
[0025]
根据本发明的一些实施方式,步骤s2中所述无机碱为碳酸钠、碳酸钾或磷酸钾中至少一种。
[0026]
根据本发明的一些实施方式,所述溶剂为冰醋酸。
[0027]
根据本发明的一些实施方式,所述有机碱为醋酸钾或醋酸钠。
[0028]
根据本发明的一些实施方式,所述在酸性条件下是指将溶剂的ph值调成小于7即可。包括但不限于在溶剂中加入盐酸、硫酸或硝酸。
[0029]
本发明的第三方面提供了一种酰胺类化合物在制备krasg12c降解剂或在抗癌药物中的应用。
[0030]
本发明的第四方面还提供了一种药物组合物,所述组合物含有本发明的酰胺类化合物或其药学上可接受的盐,和药学上可接受的赋形剂。
[0031]
一般术语
[0032]
本发明中所述“药学上可接受的”是指在施用于动物或人类时不产生不良反应、过敏反应或其他不利反应的分子实体和组合物,如本发明中的“药学上可接受的赋形剂”包括任何和所有溶剂、分散介质、包衣、抗细菌剂和抗真菌剂、等渗剂和吸收延迟剂等,此类赋形剂用于药学活性物质的使用是本领域所熟知的。
[0033]
本发明中所述“药学上可接受的盐”包括碱加成盐和酸加成盐。
[0034]
药学上可接受的碱加成盐可以用金属或胺(例如碱金属和碱土金属或有机胺)来形成。化合物的药学上可接受的盐也可以用药学上可接受的阳离子来制备。适合的药学上可接受的阳离子是本领域技术人员所熟知的并且包括碱金属阳离子、碱土金属阳离子、铵阳离子和季铵阳离子。碳酸盐或碳酸氢盐也是可能的。用作阳离子的金属的是钠、钾、镁、铵、钙或三价铁等。适合的胺的包括异丙胺、三甲胺、组氨酸、n,n'-二苄基乙二胺、氯普鲁卡因、胆碱、二乙醇胺、二环己胺、乙二胺、n-甲基葡糖胺和普鲁卡因。
[0035]
药学上可接受的酸加成盐包括无机酸盐或有机酸盐。适合的酸盐包括盐酸盐、甲酸盐、乙酸盐、柠檬酸盐、水杨酸盐、硝酸盐、磷酸盐。其他适合的药学上可接受的盐是本领域技术人员所熟知的并且包括例如甲酸、乙酸、柠檬酸、草酸、酒石酸或扁桃酸、盐酸、氢溴酸、硫酸或磷酸;与有机羧酸、磺酸、磺酸基酸或磷酸基酸或n-取代的氨基磺酸,例如乙酸、三氟乙酸(tfa)、丙酸、乙醇酸、琥珀酸、马来酸、羟基马来酸、甲基马来酸、富马酸、苹果酸、酒石酸、乳酸、草酸、葡糖酸、葡糖二酸、葡糖醛酸、柠檬酸、苯甲酸、肉桂酸、扁桃酸、水杨酸、
4-氨基水杨酸、2-苯氧基苯甲酸、2-乙酰氧基苯甲酸、扑酸、烟酸或异烟酸的盐;以及与氨基酸,例如在自然界中参与蛋白质合成的20种α氨基酸,例如谷氨酸或天冬氨酸,以及与苯乙酸、甲磺酸、乙磺酸、2-羟基乙磺酸、乙烷1,2-二磺酸、苯磺酸、4-甲基苯磺酸、萘2-磺酸、萘1,5-二磺酸、2-磷酸甘油酸或3-磷酸甘油酸、葡萄糖6-磷酸、n-环己基氨基磺酸(用于环己氨磺酸盐的形成),或与其他酸性有机化合物,例如抗坏血酸的盐。
[0036]
含有本发明所述的药物组合物能以常规方式来制造,例如通过常规混合、溶解、造粒、糖衣丸制备、磨细、乳化、囊封、捕集或冻干方法。适当的配制品取决于所选的施用途径。
附图说明
[0037]
图1是实施例1制备的酰胺类化合物对krasc12c的降解的浓度和时间关系图;
[0038]
图2是实施例1制备的化合物、对比例1的kras g12c-in-3和对比例2的pomalidomide降解效果图;
[0039]
图3是实施例1制备的酰胺类化合物对降解krasc12c和krasg13d的效果图;
[0040]
图4实施例1制备是不同浓度的酰胺类化合物对抑制krasg12c突变的h358肿瘤细胞图;
[0041]
图5是实施例1制备的酰胺类化合物的核磁共振氢谱图;
[0042]
图6是实施例1制备的酰胺类化合物的质谱图。
[0043]
图7是对比例2的核磁共振氢谱图。
具体实施方式
[0044]
如无特殊说明,本发明所用原料、试剂及溶剂,均为商业购买未经任何处理或者可通过文献方法制得。为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
[0045]
rpmi-1640培养基购自gibco;胎牛血清购自capricorn scientific;二甲基亚砜(dmso)、四甲基偶氮唑蓝(mtt)均购自sigma。细胞购买自长沙优泽生物科技有限公司,所购细胞均经str鉴定。
[0046]
实施例1
[0047]
实施例1提供了一种酰胺类化合物,所述酰胺类化合物的结构式如式(ⅰ)所示:
[0048][0049]
其制备方法包括如下步骤:
[0050]
化合物1的制备:
[0051]
将167mg 3-氟磷酸酐和164mg 3-氨基-2,6-哌啶二酮溶解于5ml冰醋酸中,加入醋酸钾82mg,80度回流过夜,加水析出得到化合物1;
[0052]
s1.将化合物1(276mg)、boc-己二胺(167mg)和n,n-二异丙基乙胺(90mg)加入到n,n-二甲基甲酰胺中进行反应得到化合物2,化合物2在盐酸条件下脱除boc,旋干溶剂得到化合物3;
[0053]
化合物4的制备:
[0054]
将4-氯-5-碘-2-甲氧基苯甲酸(311mg)和boc哌嗪(186mg)溶解在dmf中,加入dipea(90mg)在90℃反应,加水析出得到化合物4。
[0055]
s2.在n,n-二甲基甲酰胺中加入化合物4(480mg)和2,5-二氯苯硼酸(190mg),在四三苯基膦钯7mg和碳酸钠300mg的条件下进行反应,通过柱层析法得到化合物5;化合物5在盐酸条件下脱除boc,得到化合物6;
[0056]
s3.将化合物6(368mg)、顺丁烯二酸酐(98mg)和n,n-二异丙基乙胺(90mg)在n,n-二甲基甲酰胺中反应得到化合物7;
[0057]
s4.将化合物7(497mg)、化合物3(316mg)、1-羟基苯并三唑(440mg)和三乙胺(90mg)在n,n-二甲基甲酰胺中反应得到式(ⅰ)化合物;
[0058]
所述化合物1~7的结构式如下:
[0059][0060]
将所得到产物用核磁共振确认结构:
[0061]
n-(5-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)pentyl)-4-oxo-4-(4-(2',5',6-trichloro-4-methoxy-[1,1'-biphenyl]-3-carbonyl)piperazin-1-yl)but-2-enamide(式(ⅰ)化合物):yellow oil.yield:12%.1h nmr(400mhz,cdcl3)δ8.34(d,j=7.4hz,1h),7.51(t,j=7.2hz,1h),7.42(d,j=8.5hz,1h),7.36
–
7.29(m,2h),7.17(s,1h),7.09(dd,j=13.0,7.4hz,2h),6.90(d,j=8.6hz,1h),6.35(dd,j=20.5,12.4hz,1h),6.24(s,1h),6.09(d,j=12.3hz,1h),4.93(m,1h),3.91(s,3h),3.71(m,3h),3.57(m,1h),3.48
–
3.22(m,7h),2.93
–
2.70(m,3h),2.15(m,1h),1.60
–
1.53(m,2h),1.41(m,4h).ms calculated for c
41h41
cl3n6o8na[m+na]
+
873.9,found 873.9.ir(kbr)νmax=3387,2921,2855,1698,1616,1451,1360,1256,1189,1100,1016,844,742cm-1
.
[0062]
对比例1
[0063]
对比例1为kras g12c-in-3,结构式为
[0064]
对比例2
[0065]
对比例2为pomalidomide(3-氨基-n-(2,6-二氧代-3-哌啶基)邻苯二甲酰亚胺),结构式为购自invivochem。
[0066]
性能测试
[0067]
应用例1
[0068]
将实施例1制备的酰胺类化合物与对比例1的kras g12c-in-3和对比例2的pomalidomide进行krasg12c降解效果研究;具体测试方法如下:
[0069]
1.细胞总蛋白提取
[0070]
(1)每40ul ripa裂解液中加入0.4ul pmsf(100x)和0.4ul磷酸酶抑制剂(100x)混
匀。
[0071]
(2)培养细胞弃培养基,pbs清洗,加入40ul ripa裂解液吹打后转移到1.5ml ep管中。
[0072]
(3)1.5mlep管冰上摇30min,4℃12000g离心20min,取上清转移到新的1.5ml ep管中。
[0073]
2.bca法测定总蛋白浓度
[0074]
3.4x loading buffer用蛋白样品稀释至1x loading buffer,100℃变性5min,分装到1.5ml ep管中,直接使用或-40℃保存。最终蛋白上样量为20-50ug;
[0075]
4.sds-page配制:根据目的蛋白的分子量使用12%分离胶和浓缩胶(5%)。
[0076]
5.电泳:向电泳槽中倒入4℃过夜预冷的1x tris-gly电泳缓冲液。按照等质量等体积原则向每个泳道中加入蛋白样品,两侧加入3ul蛋白marker。80v恒压电泳20min,提高电压至120v,继续电泳直至溴酚蓝到达分离胶底部,约90min。
[0077]
6.转膜:准备海绵、滤纸、pvdf膜,将4℃过夜预冷的1x转移缓冲液和甲醇分别倒入搪瓷盘和孵育盒中,pvdf膜置于甲醇中浸泡3min后再浸泡到转移液中,海绵、滤纸置于转膜液中充分浸泡。根据目的蛋白大小,切去多余分离胶,放置于转膜液中。将转移槽放入装满碎冰的泡沫盒中。接通电源,100v恒压转移75min或300ma恒流转移75min将pvdf膜从封闭液中转移到tbst中,用tbst漂洗5遍,每次5min,然后按照目的蛋白分子量大小裁取条带。
[0078]
7.封闭:配制5%脱脂奶粉,充分混匀后倒入孵育盒中,摇床上缓慢摇动封闭1h。
[0079]
8.配制一抗反应液:将条带放入装有一抗反应液的离心管中,4℃振荡孵育过夜。
[0080]
9.tbst洗5次,每次10min。
[0081]
10.配制二抗反应液:根据抗体说明书标注的稀释比例用1xtbst配置抗体,颠倒混匀,冰上放置。将条带放入装有二抗反应液的离心管中,室温振荡孵育1h。1xtbst洗5次,每次10min。
[0082]
11.曝光。
[0083]
根据上述体外实验结果,图1是实施例1制备的酰胺类化合物对krasc12c的降解的浓度和时间关系图;我们可以得出从图1看出,本技术实施例1制备的化合物能够降解krasg12c且成浓度依赖性和时间依赖性;图2是实施例1制备的化合物、对比例2kras g12c-in-3和对比例3的pomalidomide降解效果图;从图2可以看出,本技术化合物对krasg12c的降解效果明显优于对比例1和对比例2;图3是实施例1制备的酰胺类化合物对降解krasc12c和krasg13d的效果图;从图3可以看出,本技术制备化合物能选择性降解h358和h23细胞中的krasg12c而不降解hct116细胞中的krasg13d。
[0084]
应用例2
[0085]
本技术实施例1制备的酰胺类化合物、对比例1和对比例2的化合物对h358肺癌细胞抗肿瘤活性研究,采用mtt法检测化合物对肿瘤细胞增殖的抑制效果。
[0086]
实验原理:mtt比色法是一种检测细胞存活和生长的方法,其原理为活细胞线粒体中的琥珀酸脱氢酶能使外源性mtt还原为水不溶性蓝紫色结晶甲瓒,并沉积在细胞中,而死细胞缺少这一功能。二甲基亚砜(dmso)可以溶解活细胞中的甲瓒,用酶联免疫检测仪检测570nm下的吸光度值(od值),可以根据吸光度值反应活细胞的数量,在一定范围内,od值越小,则表明细胞活性越弱,药物的抑制增殖效果越好。
[0087]
试剂配制
[0088]
1、mtt
[0089]
将50ml的离心管用锡箔纸包裹避光,精密称取mtt粉末250mg,加入到离心管中,加入50ml的pbs,使mtt粉末完全溶解,用0.22μm孔径的滤膜过滤除菌并分装,在-20℃条件下避光保存。
[0090]
2、实施例1化合物配置
[0091]
取高压好的ep管用于称取化合物,向ep管中加入对应量的dmso,使液体成20mm的母液,使用时用相应量的培养基稀释1配成浓度为5μm,10μm,25μm,50μm,100μm工作液。
[0092]
实验步骤:
[0093]
(1)取对数生长期的细胞,消化,调整细胞数浓度为2.5
×
104/ml,按100μl/孔接种到96孔板中。在37℃,5%co2细胞培养箱中培养过夜,待细胞贴壁。
[0094]
(2)吸出原有培养基,每组加入不同浓度的实施例1化合物,化合物浓度分别为5μm,10μm,25μm,50μm,100μm。以0.1%的dmso设为对照组,在细胞培养箱中继续培养48h。
[0095]
(3)每孔加入10μl mtt液,在培养箱中孵育4h。
[0096]
(4)弃去培养基,每孔加入100μl dmso,振荡15min充分溶解甲瓒结晶。
[0097]
(5)用酶联免疫检测仪测定570nm下的吸光度值。
[0098]
(6)按以下公式计算细胞生长抑制率:
[0099]
抑制率=[(as-ab)/(ac-ab)]
×
100%;
[0100]
as:实验孔的吸光度(含细胞、mtt、实施例1化合物);
[0101]
ac:对照孔的吸光度(含细胞、mtt,无实施例1化合物);
[0102]
ab:空白孔的吸光度(不含细胞和实施例1化合物,含mtt);
[0103]
同上述方法,分别用对比例1和对比例2的化合物其替代本技术实施例1的化合物再测试两次。
[0104]
根据上述试验,得出来的数据如表1所示:
[0105]
表1实施例和对比例的数据
[0106] h358细胞(ic
50
)本技术实施例1制备的化合物17.41
±
0.45μm对比例1》50μm对比例2》100μm
[0107]
根据上述体外实验结果,从图4可以看出,实施例1化合物能有效抑制抑制h358细胞生长,ic
50
为17.41
±
0.45μm。平板形成实验证明实施例1化合物浓度在5μm,10μm,15μm,20μm时能有效抑制h358细胞的生长。我们可以得出本技术所述的实施例1化合物能够抑制krasg12c突变的h358肿瘤细胞生长且活性明显优于对比例1的kras g12c-in-3和对比例2的pomalidomide。
[0108]
上面结合实施例对本发明作了详细说明,但是本发明不限于上述实施例,在所属技术领域普通技术人员所具备的知识范围内,还可以在不脱离本发明宗旨的前提下作出各种变化。