1.本发明涉及药物技术领域,更具体地说,是涉及一种2-((2-甲氧基苯基)磺酰基)异吲哚啉类化合物及其制备方法。
背景技术:
2.rho属于小分子单聚体gtpase超家族,是ras超家族的哺乳动物基因同系物,通过下游最主要的效应分子rho激酶来调节细胞肌动蛋白骨架的重组,从而广泛参与细胞有丝分裂、细胞骨架调整、神经再生、肿瘤细胞浸润、细胞凋亡等一系列生物学过程。目前发现rho激酶主要有两个亚型:rock1和rock2,前者主要存在于非神经组织如心脏、肺等,后者主要存在于中枢神经系统如神经元、大脑皮质等。
3.在多种心血管疾病中发现rho异常活化,如动脉粥样硬化、高血压、肺动脉高压等。在动物模型中rho抑制剂展现出治疗这些疾病的益处。另外,有证据表明抑制rho激酶在体内的激活还具有促进神经突触生长、促进损伤后的神经功能恢复的效果,可以治疗和缓解脊髓损伤、阿尔茨海默病、神经炎症脊髓脱鞘等中枢神经性疾病。除此之外,肿瘤的浸润和迁移也依赖于rho激酶的激活,抑制rho激酶的激活还能抑制肿瘤的转移。
4.目前已上市的rock抑制剂只有eril(用于治疗脑血管痉挛)和glanatec(用于治疗高眼压症和青光眼)。因此开发结构新颖、活性较强的rock抑制剂具有重要意义。
技术实现要素:
5.本发明为了寻找新型的rock抑制剂,经过广泛深入的研究,设计、合成了一系列结构新颖、对rho激酶的抑制活性较高的2-((2-甲氧基苯基)磺酰基)异吲哚啉类化合物,并且研究了这一类化合物的rock抑制活性。
6.本发明的一个目的是在于提供一种2-((2-甲氧基苯基)磺酰基)异吲哚啉类化合物或其药学上可接受的盐。
7.本发明的另一个目的是提供一种上述化合物的制备方法。
8.本发明的另一个目的是提供一种包含上述化合物的药物组合物。
9.本发明的另一个目的是提供上述化合物和上述药物组合物作为rho激酶抑制剂用于治疗肿瘤、心血管疾病、神经系统疾病、纤维化疾病的用途。
10.为实现上述发明目的,本发明采用的技术方案是:第一方面,本发明提供一种具有式ⅰ结构的化合物或其药学上可接受的盐:其中,r1为甲氧基、氟、氯、溴、氰基、氨基羰基、乙酰基、二甲基氨基、三氟甲基,r2为1h-吲唑-5-基、1h-吡唑-4-基。
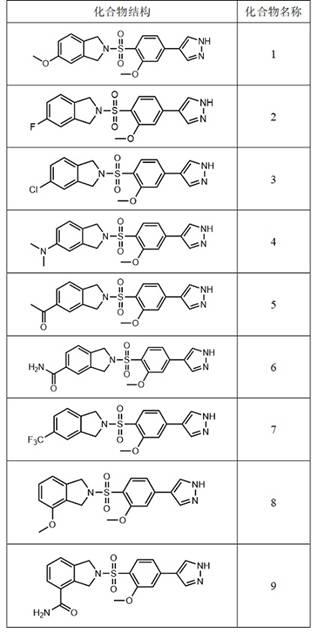
11.本发明提供的具有式ⅰ结构的化合物或其药学上可接受的盐,可以选自具有如下化合物结构的化合物:
。
12.第二方面,本发明提供了一种具有式ⅰ结构的化合物或其药学上可接受的盐的制备方法,包括如下步骤:中间体ⅳ的合成:具有式ⅱ结构的化合物ⅱ和具有式ⅲ结构的化合物ⅲ于第一种碱存在的第一溶剂中,在第一反应温度下发生反应生成具有式ⅳ结构的中间体ⅳ;
化合物ⅰ的合成:中间体ⅳ和具有式
ⅴ
结构的化合物
ⅴ
在催化剂的作用下于第二种碱存在的第二溶剂中,在第二反应温度下发生偶联反应生成具有式ⅰ结构的化合物。
13.作为本发明所提供的制备方法的一个优选方案,所述第一溶剂为四氢呋喃、二氧六环、甲苯、n,n-二甲基甲酰胺、二氯甲烷、乙腈中的至少一种;和/或,所述第二溶剂为二氧六环、n,n-二甲基甲酰胺、甲苯、二氧六环/水、乙二醇二甲醚中的任意一种。
14.作为本发明所提供的制备方法的另一个优选方案,所述第一反应温度为20-100℃;和/或,所述第二反应温度为40-120℃。
15.作为本发明所提供的制备方法的另一个优选方案,所述第一种碱为三乙胺、n,n-二异丙基乙胺、n-甲基吗啉、碳酸钾、碳酸铯、碳酸钠中的至少一种;和/或,所述第二种碱为碳酸铯、叔丁醇钠、叔丁醇钾、碳酸钾、碳酸钠、乙酸钠、磷酸钾中的至少一种。
16.作为本发明所提供的制备方法的另一个优选方案,所述催化剂为四三苯基膦钯(pd(pph3)4)、醋酸钯(pd(oac)2)、三二亚苄基丙酮二钯(pd2(dba)3)、1,1'-双(二苯膦基)二茂铁二氯化钯(pd(dppf)cl2)中的至少一种本发明提供的制备方法以化合物ⅱ和化合物ⅲ为起始原料,先合成中间体ⅳ,再通过中间体ⅳ和化合物
ⅴ
发生偶联反应制备出具有式ⅰ结构的化合物。整个制备过程操作简单,易于控制,对生产设备要求不高,适用于工业化大规模生产。
17.第三方面,本发明提供了一种药物组合物,包含具有式ⅰ结构的化合物、或其药学上可接受的盐。
18.在本发明中,具有式ⅰ结构的化合物或其药学上可接受的盐是药物组合物中的药物活性成分。
19.本发明提供的药物组合物除了包含治疗有效量的至少一种具有式ⅰ结构的化合物或其药学上可接受的盐之外,还包含:一种或多种药用辅料;和/或,一种或多种除了具有式ⅰ结构的化合物或其药学上可接受的盐之外的其它具有rock抑制活性的药用活性物质。
20.制备本发明提供的药物组合物的方法对本领域技术人员而言是显而易见的,包括常见的混合、溶解、冻干等技术。
21.本发明提供的药物组合物可以依照药学领域的常规制备方法制成各种常见的剂型,例如片剂、丸剂、胶囊剂、颗粒剂、口服溶液剂、口服混悬剂、口服乳剂、注射剂等。方便提供给患者临床使用,通过各种常见的给药方式向患者施用,例如口服或肠胃外施用(通过静脉内、肌内、局部或皮下途径)。
22.第四方面,本发明提供了具有式ⅰ结构的化合物、其药学上可接受的盐和包含具有式ⅰ结构的化合物、其药学上可接受的盐的药物组合物作为rock抑制剂在制备治疗肿瘤、心血管疾病、神经系统疾病、纤维化疾病的药物中的应用。
23.进一步的,所述肿瘤选自:
皮肤癌、膀胱癌、卵巢癌、乳腺癌、胃癌、前列腺癌、结肠癌、肺癌、骨癌、脑癌、直肠癌、食管癌、舌癌、肾癌、宫颈癌、子宫体癌、睾丸癌、泌尿癌、黑素癌、星型细胞癌、脑膜瘤、霍奇金淋巴瘤、非霍奇金淋巴瘤、急性淋巴性白血病、慢性淋巴性白血病、急性骨髓性白血病、慢性粒细胞白血病、成人t细胞白血病淋巴瘤、肝细胞癌、多发性骨髓瘤、基底细胞瘤、精原细胞瘤、软骨肉瘤、肌肉瘤、纤维肉瘤。
24.本发明的表达中涉及的一些术语定义如下:术语“药学上可接受的盐”是指那些保留母体化合物的生物有效性及特性的盐。该盐包括:酸加成盐,其是通过母体化合物的游离碱与无机酸或与有机酸的反应而获得的;所述无机酸包括盐酸、氢溴酸、氢碘酸、硝酸、磷酸、硫酸及高氯酸等;所述有机酸包括乙酸、草酸、(d)或(l)苹果酸、马来酸、甲烷磺酸、乙烷磺酸、对甲苯磺酸、水杨酸、酒石酸、苯磺酸、苯甲酸、樟脑磺酸、柠檬酸、富马酸、葡萄糖酸、谷氨酸、羟乙磺酸、乳酸、扁桃酸、黏液酸、双羟萘酸、泛酸、琥珀酸或丙二酸等;优选为盐酸或(l)-苹果酸;或者,当母体化合物中存在的酸质子被置换为金属离子或与有机碱配位时形成的盐;所述金属离子包括碱金属离子、碱土离子、铝离子等;所述有机碱包括乙醇胺、二乙醇胺、三乙醇胺、缓血酸胺、n-甲基葡糖胺等。
25.术语“水合物”是指水分子以配位键或共价键与化合物中的阳离子或阴离子结合,或指水离子不直接与阳离子或阴离子结合而是以一定比例存在于固体晶格的确定位置而形成的物质。
26.术语“溶剂合物”是指化合物与药学上可接受的溶剂分子缔合形成的物质,药学上可接受的溶剂一般包括乙醇、乙酸等。
27.术语“立体异构体”是指具有相同分子式、但是分子中原子在空间排列方式上不同的化合物。由于本发明提供的化合物可具有一个或多个不对称中心,因此该化合物可以单独(r)-立体异构体形式制备或以单独(s)-立体异构体形式制备或以其混合物形式制备。除非另有说明,否则本发明中的特定化合物的描述或名称意欲包括个别对映异构体与其外消旋混合物或其它混合物。用于测定立体化学构型及分离立体异构体的方法是本领域的常规技术(参见《ad
ⅴ
anced organic chemistry》的第4章中的论述,第4版,j.march,john wiley及sons,new york,1992)。因此,本发明亦涵盖具有调节ret激酶活性的能力的任何立体异构形式、其相应对映异构体(d-异构体及l-异构体或(+)异构体及(-)异构体)及其非对映异构体及其混合物且不限于任一种立体异构形式。
28.术语“药物组合物”是指一种或多种本发明所提供的化合物与其它化学成分(例如药用辅料)的混合物。药物组合物的目的旨在促进化合物给予生物。
29.术语“药用辅料”是指除活性成分外,在安全性方面已进行了合理的评估,对生物不产生明显刺激且不会消除所给予的化合物的生物活性及特性,并且包含在药物制剂中的物质。药用辅料除了赋型、充当载体、提高稳定性外,还具有增溶、助溶、缓控释等重要功能,是可能会影响到药物组合物的质量、安全性和有效性的重要成分。药用辅料包括但不限于载体、稀释剂、赋形剂、增溶剂、助溶剂、黏合剂、崩解剂、渗透促进剂、ph调节剂、缓冲剂、释放阻滞剂、矫味剂、防腐剂、抗氧剂等。
30.本发明的有益效果至少在于:
(1)本发明提供了一类2-((2-甲氧基苯基)磺酰基)异吲哚啉类化合物或其药学上可接受的盐,可以作为有效的rock抑制剂,其抑制活性较强。
31.(2)本发明还提供了一种包含2-((2-甲氧基苯基)磺酰基)异吲哚啉类化合物或其药学上可接受的盐的药物组合物,具有很好的治疗肿瘤、心血管疾病、神经系统疾病、纤维化疾病的药理活性。
具体实施方式
32.为了使本发明要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合具体的实施方式,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施方式只是用于详细说明本专利,并不以任何方式限制本发明的保护范围。
33.在本发明的具有式ⅰ结构的化合物或其药学上可接受的盐的制备方法中,具体反应过程如下:第一步要合成中间体ⅳ:将化合物ⅱ、化合物ⅲ和第一种碱溶于第一溶剂中,在20-100℃下发生反应生成中间体ⅳ。
34.其中,第一种碱可以选择三乙胺、n,n-二异丙基乙胺、n-甲基吗啉、碳酸钾、碳酸铯、碳酸钠中的至少一种。
35.第一溶剂可以选择四氢呋喃、二氧六环、甲苯、n,n-二甲基甲酰胺、二氯甲烷、乙腈中的至少一种。
36.例如,将1mol化合物ⅱ、1mol化合物ⅲ和1.5mol n,n-二异丙基乙胺溶于5l四氢呋喃中,40℃下搅拌反应生成中间体ⅳ。可以采用薄层色谱(thin layer chromatography,简写为tlc)监测反应,反应完毕后经提取、干燥、浓缩、分离等常规处理工艺即可得到中间体ⅳ。
37.第二步是合成具有式ⅰ结构的化合物:将中间体ⅳ、化合物
ⅴ
、第二种碱和催化剂溶于第二溶剂中,在40-120℃下发生偶联反应生成具有式ⅰ结构的化合物。
38.其中,第二种碱可以选择碳酸铯、叔丁醇钠、叔丁醇钾、碳酸钾、碳酸钠、乙酸钠、磷酸钾中的至少一种。
39.第二溶剂可以选择二氧六环、n,n-二甲基甲酰胺、甲苯、二氧六环/水、乙二醇二甲
醚中的任意一种。
40.催化剂可以选择四三苯基膦钯(pd(pph3)4)、醋酸钯(pd(oac)2)、三二亚苄基丙酮二钯(pd2(dba)3)、1,1'-双(二苯膦基)二茂铁二氯化钯(pd(dppf)cl2)中的至少一种。
41.例如,将1mol中间体ⅳ、1mol化合物
ⅴ
、2mol乙酸钠和0.1mol醋酸钯(pd(oac)2)溶于4l二氧六环中,在75℃搅拌反应生成具有式ⅰ结构的化合物。可以采用tlc监测反应,反应完毕以后,再经过浓缩、分离等常规处理工艺即可获得最终产物。
42.本发明先后进行多次试验,现列举一部分试验作为参考对发明进行进一步详细描述,下面结合具体实施例进行详细说明:除有定义外,以下实施例中所用的技术术语具有与本发明所属领域技术人员普遍理解的相同含义。以下实施例中所用的试剂,如无特殊说明,均为常规生化试剂;以下实施例中所用的原材料、仪器和设备等,均可通过市场购买获得或者可通过现有方法获得;所述试剂用量,如无特殊说明,均为常规实验操作中试剂用量;所述实验方法,如无特殊说明,均为常规方法。
43.实施例15-甲氧基-2-((2-甲氧基-4-(1h-吡唑-4-基)苯基)磺酰基)异吲哚啉第一步:将化合物1a(2.84g,10.0mmol)、化合物1b(1.5g,10.0mmol)、n,n-二异丙基乙胺(diea)(1.9g,15.0mmol)溶于二氯甲烷(50ml)中,室温反应,tlc监测反应,反应完毕后加水(50ml)淬灭反应,有机层干燥,过滤、浓缩、柱层析分离得到淡黄色固体(化合物1c)2.6g,收率65.5%;第二步:将化合物1c(397mg,1.0mmol)、化合物1d(112mg,1.0mmol)、碳酸钾(276mg,2.0mmol)、四三苯基膦钯(pd(pph3)4)(115mg,0.1mmol)溶于二氧六环(30ml)和水(10ml),升温至80℃搅拌反应,tlc监测反应,反应完毕后用乙酸乙酯(50ml
×
2)提取,有机层浓缩,柱层析分离得到类白色固体(化合物1)224mg,收率为58.2%。esi(+)m/z=386.1。
44.实施例25-氟-2-((2-甲氧基-4-(1h-吡唑-4-基)苯基)磺酰基)异吲哚啉
第一步:将化合物1a(2.84g,10.0mmol)、化合物2a(1.37g,10.0mmol)、diea(1.9g,15.0mmol)溶于二氯甲烷(50ml)中,室温反应,tlc监测反应,反应完毕后加水(50ml)淬灭反应,有机层干燥,过滤、浓缩、柱层析分离得到淡黄色固体(化合物2b)2.7g,收率70.3%;第二步:将化合物2b(385mg,1.0mmol)、化合物1d(112mg,1.0mmol)、碳酸钾(276mg,2.0mmol)、四三苯基膦钯(115mg,0.1mmol)溶于二氧六环(30ml)和水(10ml),升温至80℃搅拌反应,tlc监测反应,反应完毕后用乙酸乙酯(50ml
×
2)提取,有机层浓缩,柱层析分离得到类白色固体(化合物2)257mg,收率为68.9%。esi(+)m/z=374.1。
45.实施例35-氯-2-((2-甲氧基-4-(1h-吡唑-4-基)苯基)磺酰基)异吲哚啉第一步:将化合物1a(2.84g,10.0mmol)、化合物3a(1.5g,10.0mmol)、diea(1.9g,15.0mmol)溶于二氯甲烷(50ml)中,室温反应,tlc监测反应,反应完毕后加水(50ml)淬灭反应,有机层干燥,过滤、浓缩、柱层析分离得到淡黄色固体(化合物3b)2.2g,收率55.0%;第二步:
将化合物3b(400mg,1.0mmol)、化合物1d(112mg,1.0mmol)、碳酸钾(276mg,2.0mmol)、四三苯基膦钯(115mg,0.1mmol)溶于二氧六环(30ml)和水(10ml),升温至80℃搅拌反应,tlc监测反应,反应完毕后用乙酸乙酯(50ml
×
2)提取,有机层浓缩,柱层析分离得到类白色固体(化合物3)209mg,收率为53.6%。esi(+)m/z=390.1。
46.实施例45-二甲基氨基-2-((2-甲氧基-4-(1h-吡唑-4-基)苯基)磺酰基)异吲哚啉第一步:将化合物1a(2.84g,10.0mmol)、化合物4a(1.6g,10.0mmol)、diea(1.9g,15.0mmol)溶于二氯甲烷(50ml)中,室温反应,tlc监测反应,反应完毕后加水(50ml)淬灭反应,有机层干燥,过滤、浓缩、柱层析分离得到淡黄色固体(化合物4b)1.9g,收率46.3%;第二步:将化合物4b(410mg,1.0mmol)、化合物1d(112mg,1.0mmol)、碳酸钾(276mg,2.0mmol)、四三苯基膦钯(115mg,0.1mmol)溶于二氧六环(30ml)和水(10ml),升温至80℃搅拌反应,tlc监测反应,反应完毕后用乙酸乙酯(50ml
×
2)提取,有机层浓缩,柱层析分离得到类白色固体(化合物4)252mg,收率为63.3%。esi(+)m/z=399.1。
47.实施例55-乙酰基-2-((2-甲氧基-4-(1h-吡唑-4-基)苯基)磺酰基)异吲哚啉
第一步:将化合物1a(2.84g,10.0mmol)、化合物5a(1.6g,10.0mmol)、diea(1.9g,15.0mmol)溶于二氯甲烷(50ml)中,室温反应,tlc监测反应,反应完毕后加水(50ml)淬灭反应,有机层干燥,过滤、浓缩、柱层析分离得到淡黄色固体(化合物5b)2.5g,收率61.1%;第二步:将化合物5b(409mg,1.0mmol)、化合物1d(112mg,1.0mmol)、碳酸钾(276mg,2.0mmol)、四三苯基膦钯(115mg,0.1mmol)溶于二氧六环(30ml)和水(10ml),升温至80℃搅拌反应,tlc监测反应,反应完毕后用乙酸乙酯(50ml
×
2)提取,有机层浓缩,柱层析分离得到类白色固体(化合物5)271mg,收率为68.3%。esi(+)m/z=398.1。
48.实施例65-氨基羰基-2-((2-甲氧基-4-(1h-吡唑-4-基)苯基)磺酰基)异吲哚啉第一步:将化合物1a(2.84g,10.0mmol)、化合物6a(1.6g,10.0mmol)、diea(1.9g,15.0mmol)溶于二氯甲烷(50ml)中,室温反应,tlc监测反应,反应完毕后加水(50ml)淬灭反应,有机层干燥,过滤、浓缩、柱层析分离得到淡黄色固体(化合物6b)2.2g,收率53.8%;第二步:将化合物6b(409mg,1.0mmol)、化合物1d(112mg,1.0mmol)、碳酸钾(276mg,2.0mmol)、四三苯基膦钯(115mg,0.1mmol)溶于二氧六环(30ml)和水(10ml),升温至80℃搅拌反应,tlc监测反应,反应完毕后用乙酸乙酯(50ml
×
2)提取,有机层浓缩,柱层析分离得到白色固体(化合物6)285mg,收率为71.6%。esi(+)m/z=399.1。
49.实施例75-三氟甲基-2-((2-甲氧基-4-(1h-吡唑-4-基)苯基)磺酰基)异吲哚啉
第一步:将化合物1a(2.84g,10.0mmol)、化合物7a(1.9g,10.0mmol)、diea(1.9g,15.0mmol)溶于二氯甲烷(50ml)中,室温反应,tlc监测反应,反应完毕后加水(50ml)淬灭反应,有机层干燥,过滤、浓缩、柱层析分离得到淡黄色固体(化合物7b)2.6g,收率59.8%;第二步:将化合物7b(434mg,1.0mmol)、化合物1d(112mg,1.0mmol)、碳酸钾(276mg,2.0mmol)、四三苯基膦钯(115mg,0.1mmol)溶于二氧六环(30ml)和水(10ml),升温至80℃搅拌反应,tlc监测反应,反应完毕后用乙酸乙酯(50ml
×
2)提取,有机层浓缩,柱层析分离得到白色固体(化合物7)225mg,收率为53.2%。esi(+)m/z=424.1。
50.实施例84-甲氧基-2-((2-甲氧基-4-(1h-吡唑-4-基)苯基)磺酰基)异吲哚啉第一步:将化合物1a(2.84g,10.0mmol)、化合物8a(1.5g,10.0mmol)、diea(1.9g,15.0mmol)溶于二氯甲烷(50ml)中,室温反应,tlc监测反应,反应完毕后加水(50ml)淬灭反应,有机层干燥,过滤、浓缩、柱层析分离得到淡黄色固体(化合物8b)2.8g,收率70.5%;第二步:将化合物8b(397mg,1.0mmol)、化合物1d(112mg,1.0mmol)、碳酸钾(276mg,
2.0mmol)、四三苯基膦钯(115mg,0.1mmol)溶于二氧六环(30ml)和水(10ml),升温至80℃搅拌反应,tlc监测反应,反应完毕后用乙酸乙酯(50ml
×
2)提取,有机层浓缩,柱层析分离得到白色固体(化合物8)248mg,收率为64.4%。esi(+)m/z=386.1。
51.实施例94-氨基羰基-2-((2-甲氧基-4-(1h-吡唑-4-基)苯基)磺酰基)异吲哚啉第一步:将化合物1a(2.84g,10.0mmol)、化合物9a(1.6g,10.0mmol)、diea(1.9g,15.0mmol)溶于二氯甲烷(50ml)中,室温反应,tlc监测反应,反应完毕后加水(50ml)淬灭反应,有机层干燥,过滤、浓缩、柱层析分离得到淡黄色固体(化合物9b)2.3g,收率56.1%;第二步:将化合物9b(410mg,1.0mmol)、化合物1d(112mg,1.0mmol)、碳酸钾(276mg,2.0mmol)、四三苯基膦钯(115mg,0.1mmol)溶于二氧六环(30ml)和水(10ml),升温至80℃搅拌反应,tlc监测反应,反应完毕后用乙酸乙酯(50ml
×
2)提取,有机层浓缩,柱层析分离得到白色固体(化合物9)208mg,收率为52.3%。esi(+)m/z=399.1。
52.实施例104-氟-2-((2-甲氧基-4-(1h-吡唑-4-基)苯基)磺酰基)异吲哚啉第一步:
将化合物1a(2.84g,10.0mmol)、化合物10a(1.4g,10.0mmol)、diea(1.9g,15.0mmol)溶于二氯甲烷(50ml)中,室温反应,tlc监测反应,反应完毕后加水(50ml)淬灭反应,有机层干燥,过滤、浓缩、柱层析分离得到淡黄色固体(化合物10b)2.0g,收率51.9%;第二步:将化合物10b(385mg,1.0mmol)、化合物1d(112mg,1.0mmol)、碳酸钾(276mg,2.0mmol)、四三苯基膦钯(115mg,0.1mmol)溶于二氧六环(30ml)和水(10ml),升温至80℃搅拌反应,tlc监测反应,反应完毕后用乙酸乙酯(50ml
×
2)提取,有机层浓缩,柱层析分离得到白色固体(化合物10)226mg,收率为60.6%。esi(+)m/z=374.1。
53.实施例115-(3-甲氧基-4-((5-甲氧基异吲哚啉-2-基)磺酰基)苯基)-1h-吲唑按照实施例1中的第一步的方法合成化合物1c。
54.将化合物1c(397mg,1.0mmol)、化合物11a(162mg,1.0mmol)、碳酸钾(276mg,2.0mmol)、四三苯基膦钯(115mg,0.1mmol)溶于二氧六环(30ml)和水(10ml),升温至80℃搅拌反应,tlc监测反应,反应完毕后用乙酸乙酯(50ml
×
2)提取,有机层浓缩,柱层析分离得到白色固体(化合物11)255mg,收率为58.6%,esi(+)m/z=436.1。
55.实施例125-(3-甲氧基-4-((5-氟异吲哚啉-2-基)磺酰基)苯基)-1h-吲唑按照实施例2中的第一步的方法合成化合物2b。
56.将化合物2b(385mg,1.0mmol)、化合物11a(162mg,1.0mmol)、碳酸钾(276mg,2.0mmol)、四三苯基膦钯(115mg,0.1mmol)溶于二氧六环(30ml)和水(10ml),升温至80℃搅拌反应,tlc监测反应,反应完毕后用乙酸乙酯(50ml
×
2)提取,有机层浓缩,柱层析分离得到白色固体(化合物12)278mg,收率为65.7%。esi(+)m/z=424.1。
57.实施例135-(3-甲氧基-4-((5-氯异吲哚啉-2-基)磺酰基)苯基)-1h-吲唑
按照实施例3中的第一步的方法合成化合物3b。
58.将化合物3b(400mg,1.0mmol)、化合物11a(162mg,1.0mmol)、碳酸钾(276mg,2.0mmol)、四三苯基膦钯(115mg,0.1mmol)溶于二氧六环(30ml)和水(10ml),升温至80℃搅拌反应,tlc监测反应,反应完毕后用乙酸乙酯(50ml
×
2)提取,有机层浓缩,柱层析分离得到白色固体(化合物13)278mg,收率为63.3%。esi(+)m/z=440.1。
59.实施例145-(3-甲氧基-4-((5-二甲基氨基吲哚啉-2-基)磺酰基)苯基)-1h-吲唑按照实施例4中的第一步的方法合成化合物4b。
60.将化合物4b(410mg,1.0mmol)、化合物11a(162mg,1.0mmol)、碳酸钾(276mg,2.0mmol)、四三苯基膦钯(115mg,0.1mmol)溶于二氧六环(30ml)和水(10ml),升温至80℃搅拌反应,tlc监测反应,反应完毕后用乙酸乙酯(50ml
×
2)提取,有机层浓缩,柱层析分离得到白色固体(化合物14)240mg,收率为53.6%。esi(+)m/z=449.2。
61.实施例155-(3-甲氧基-4-((5-乙酰基异吲哚啉-2-基)磺酰基)苯基)-1h-吲唑按照实施例5中的第一步的方法合成化合物5b。
62.将化合物5b(409mg,1.0mmol)、化合物11a(162mg,1.0mmol)、碳酸钾(276mg,2.0mmol)、四三苯基膦钯(115mg,0.1mmol)溶于二氧六环(30ml)和水(10ml),升温至80℃搅拌反应,tlc监测反应,反应完毕后用乙酸乙酯(50ml
×
2)提取,有机层浓缩,柱层析分离得到白色固体254mg(化合物15),收率为56.8%。esi(+)m/z=448.1。
63.实施例165-(3-甲氧基-4-((5-氨基羰基异吲哚啉-2-基)磺酰基)苯基)-1h-吲唑
按照实施例6中的第一步的方法合成化合物6b。
64.将化合物6b(409mg,1.0mmol)、化合物11a(162mg,1.0mmol)、碳酸钾(276mg,2.0mmol)、四三苯基膦钯(115mg,0.1mmol)溶于二氧六环(30ml)和水(10ml),升温至80℃搅拌反应,tlc监测反应,反应完毕后用乙酸乙酯(50ml
×
2)提取,有机层浓缩,柱层析分离得到白色固体(化合物16)282mg,收率为62.8%。esi(+)m/z=449.1。
65.实施例175-(3-甲氧基-4-((5-三氟甲基异吲哚啉-2-基)磺酰基)苯基)-1h-吲唑按照实施例7中的第一步的方法合成化合物7b。
66.将化合物7b(434mg,1.0mmol)、化合物11a(162mg,1.0mmol)、碳酸钾(276mg,2.0mmol)、四三苯基膦钯(115mg,0.1mmol)溶于二氧六环(30ml)和水(10ml),升温至80℃搅拌反应,tlc监测反应,反应完毕后用乙酸乙酯(50ml
×
2)提取,有机层浓缩,柱层析分离得到白色固体(化合物17)269mg,收率为56.9%。esi(+)m/z=474.1。
67.生物学评价本实施例通过检测实施例1~17制备的化合物1~17对rock1(rock2)激酶的抑制作用,gsk269962为内控化合物。
68.实验方法:将化合物用二甲基亚砜(dmso)稀释至10个不同浓度,备用;缓冲液:40mm tris ph 7.5,20mm mgcl2,0.1%bsa,50μm dtt;加10μl 2.5x0.1μg/ml rock1(rock2)工作液进入96孔板;加入5μl 5x化合物溶液至96孔板混匀,25℃孵育10分钟;加入10μl 2.5x37.5μg/ml s6k底物和12.5μm atp混合工作液,30℃孵育60分钟;取25μl反应混合物到另外一个96孔板中,加入25μl adp-glo试剂混合,25℃孵育60分钟后终止反应;取40μl终止反应混合液,加入40μl激酶检测试剂混匀,25℃孵育40分钟;读取iuminescenece信号值,通过计算得到化合物ic
50
。测试结果如下:
从上表中可以看出,化合物1~17对rock1(rock2)激酶均有一定的抑制作用,其中,化合物1、化合物2、化合物3、化合物5、化合物6、化合物8化合物9、化合物10、化合物11、化合物12、化合物16、化合物17对rock1的ic
50
值小于10nm;化合物2、化合物3、化合物5、化合物11~15对rock2的ic
50
值小于10nm。
69.以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。