一种2-(4-氨基苯基)-5-氨基苯并咪唑的合成方法
技术领域
1.本发明涉及2-(4-氨基苯基)-5-氨基苯并咪唑技术领域,特别涉及一种2-(4-氨基苯基)-5-氨基苯并咪唑的合成方法。
背景技术:
2.2-(4-氨基苯基)-5-氨基苯并咪唑是一种用于制备聚酰亚胺类材料的重要中间体。用其制备的聚酰亚胺材料广泛应用于电子电路基板、军用防弹材料等领域,市场需求巨大。
3.2-(4-氨基苯基)-5-氨基苯并咪唑的现有的、已实现工业化的合成方法,大致分为3类,分别要用到:2,4-二硝基苯胺;或4-硝基邻苯二胺;或1,2,4-三氨基苯盐酸盐这三种关键的化工原料。
4.使用2,4-二硝基苯胺为原料的合成方法,存在以下的缺陷:这种原料及由其制备的前期中间体都属于危险易爆的(同苯环)多硝基化合物,工艺过程及原料、中间体、三废的储藏过程的风险高;而且在工艺路线的后期、进行多硝基化合物的还原时,会生成较多的黑油状偶氮副产物;该类副产物极难除净,严重影响本方法的产物纯化收率和品质。
5.使用4-硝基邻苯二胺为原料的合成方法,存在以下的缺陷:该原料的制备过程需要使用2,4-二硝基苯胺并选择性还原其2位硝基;同样存在安全风险,而且需要用到大量的特殊化学试剂如硫化物、氯化亚锡等等,污染大、成本高。近年来多家生产4-硝基邻苯二胺的生产线因为安全、环保的原因被关停,导致这种原料的市场价格急剧上涨,相应工艺路线的成本也急剧上涨。
6.使用1,2,4-三氨基苯盐酸盐为原料的合成方法,则存在以下的缺陷:由于三氨基苯的三个氨基都有相差无几的反应活性,因此在制备反应中的选择性差,目标产物的转化率偏低、副产物多;纯化时的损耗大、收率低。近年来,也有多篇采用本类方法的研究文献声称可以达到95%以上的转化率,但均需采用昂贵的4-硝基苯甲醛为第二种关键原料,成本仍然偏高,且未实现产业化。
技术实现要素:
7.本发明提供一种2-(4-氨基苯基)-5-氨基苯并咪唑的合成方法,用以通过采用极为廉价的对氨基苯甲酸和对硝基苯甲酰氯为原料,经 4步反应得到2-(4-氨基苯基)-5-氨基苯并咪唑。本发明的目标产物总收率可达80%左右;而且中间产物不含危险的(同苯环)多硝基分子结构;整条工艺路线的成本低廉、安全性高、产生的有害污染物少。
8.本发明提供一种2-(4-氨基苯基)-5-氨基苯并咪唑的合成方法,包括:
9.步骤1,制备n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯;
10.步骤2,利用步骤1中获得的n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯制备4-(4-硝基-α-亚氨基)苯甲氨基苯甲酰胺;
11.步骤3,一锅法制备2-(4-硝基)苯基-5-氨基苯并咪唑;
12.步骤4,把2-(4-硝基)苯基-5-氨基苯并咪唑用加氢催化剂和氢气进行加氢反应,得到2-(4-氨基)苯基-4-氨基苯并咪唑。
13.优选的,所述步骤1包括:先使用对硝基苯甲酰氯与对氨基苯甲酸反应得到n-(4-硝基)苯甲酰基对氨基苯甲酸;再加入氯化亚砜反应得到n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯。
14.优选的,所述步骤1还包括:
15.步骤1.1,向溶剂一中加入对氨基苯甲酸;然后于0~30℃滴加对硝基苯甲酰氯溶液溶于溶剂一中;
16.滴加完毕后,于0~30℃搅拌继续2~10小时,生成n-(4-硝基) 苯甲酰基对氨基苯甲酸在溶剂中的悬浮物。
17.步骤1.2,把步骤1.1所得的反应混合物升温至75~85℃,然后滴加氯化亚砜;滴加完毕后,继续保温75~85℃反应5~10小时;
18.然后减压浓缩除去溶剂一和过量的氯化亚砜,即得n-(4-硝基
‑ꢀ
α-氯代)苯亚甲基对氨基苯甲酰氯的粗品。
19.优选的,其中,所述步骤1.1中还包括:
20.所述溶剂一为:1,2-二氯乙烷、甲苯、二甲苯、氯苯中的一种,或它们的混合物;
21.所述溶剂一的用量为:对氨基苯甲酸的10~20倍质量比;
22.所述对硝基苯甲酰氯的用量为:对苯二胺的1.05~1.2倍当量;
23.所述对硝基苯甲酰氯溶液为:对硝基苯甲酰氯与所述溶剂一配制而成,溶剂的配置用量为:在0~30℃把对硝基苯甲酰氯全部溶解;
24.滴加对硝基苯甲酰氯溶液的滴加温度和反应温度为:0~30℃;
25.滴加对硝基苯甲酰氯溶液完毕后的保温反应时间为:2~10小时;
26.其中,所述步骤1.2中还包括:
27.所述氯化亚砜用量为:对氨基苯甲酸的2.5~3.5倍当量;
28.滴加氯化亚砜时和滴加后的保温反应温度为:75~85℃;
29.滴加氯化亚砜后的保温反应时间为:5~10小时。
30.优选的,步骤2还包括:
31.把步骤1.2所得n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯粗品与溶剂二混合、溶清;
32.把活性炭悬浮在水中,搅拌下控温1~30℃,先加入氨水;然后滴加并滴加前述配好的、n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯的溶液;
33.滴加完毕后,继续保温1~30℃搅拌反应3~6小时,过滤;
34.滤饼经水洗除去大部分的副产物氯化铵后,即得吸附在活性炭上的4-(4-硝基-α-亚氨基)苯甲氨基苯甲酰胺湿品。
35.优选的,所述步骤2中还包括:
36.所述溶剂二为:1,2-二氯乙烷、二氯甲烷、氯仿、甲苯中的一种或多种混合物;
37.所述溶剂二的用量为:n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯粗品的4~10倍质量比;
38.所述活性炭粒度为:100~300目;所述活性炭用量为:n-(4-硝基-α-氯代)苯亚甲
基对氨基苯甲酰氯的5~10倍质量比;
39.所述水的用量为:n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯粗品的25~50倍质量比;
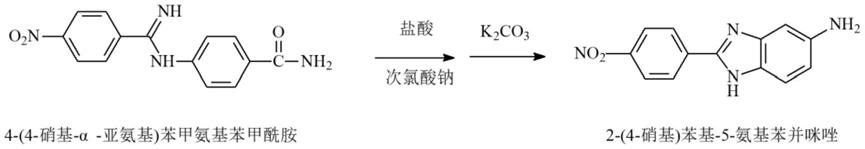
40.所述氨水的用量为:n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯的2.5~4.0倍当量;
41.其中,加入氨水、滴加n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯溶液时的温度为:1~30℃;
42.所述n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯溶液完毕后的反应温度为:1~30℃,反应时间为:3~6小时;
43.所述过滤、用水洗涤滤饼时的时的温度为0℃~室温;
44.洗涤滤饼时,洗涤至水层的ph值处于7~10。
45.优选的,所述步骤3还包括:
46.步骤3.1,把步骤2所得吸附在活性炭上的4-(4-硝基-α-亚氨基) 苯甲氨基苯甲酰胺湿品悬浮在水中,一边搅拌一边控制液温在 20~40℃,先滴加盐酸;再滴加次氯酸钠水溶液;加完后继续控制液温20~40℃搅拌1~3小时,获得悬浊液;
47.步骤3.2,把步骤3.1所得的悬浊液搅拌下降温至1~20℃,然后分批加入碳酸钾;加完后,先继续控制液温1~20℃搅拌1~3小时;再升温至至80~100℃搅拌反应8~16小时;最后降温至5~10℃,过滤;
48.所得滤饼用水洗涤后,即得吸附在活性炭上的2-(4-硝基)苯基
ꢀ‑
5-氨基苯并咪唑,其中,所述2-(4-硝基)苯基-5-氨基苯并咪唑为湿品;
49.步骤3.3,向步骤3.2所得的(水洗后的)滤饼中,加入溶剂三,并加热至60~80℃搅拌萃取1~2小时,趁热过滤;
50.步骤3.4,把步骤3.3所得滤饼,再重复一遍步骤3.2所述操作进行萃取;然后合并两次萃取的滤液,减压浓缩除去溶剂三后,即得 2-(4-硝基)苯基-5-氨基苯并咪唑粗品;
51.将2-(4-硝基)苯基-5-氨基苯并咪唑粗品用4~6倍质量的醇类溶剂四结晶提纯、再真空烘干,得2-(4-硝基)苯基-5-氨基苯并咪唑;其中,两次萃取后的滤饼为活性炭,所述活性炭可回收。
52.优选的,其中,步骤3.1还包括:
53.所述水的用量为:活性炭及4-(4-硝基-α-亚氨基)苯甲氨基苯甲酰胺湿品的总质量的3~6倍;所述盐酸的用量为:2.1中所用对氨基苯甲酸的4.0~5.0倍当量;所述次氯酸钠的用量为:2.1中所用对氨基苯甲酸的2.5~3.5倍当量;加入盐酸、次氯酸钠时的温度,及加完后搅拌时的温度均为:20~40℃;所述加完盐酸、次氯酸钠后搅拌的时间均为1~3小时;
54.所述步骤3.2还包括:
55.所述碳酸钾的用量为:2.1中所用对氨基苯甲酸的2.5~3.5倍当量;
56.所述加入碳酸钾时的温度为:1~20℃;所述加完碳酸钾后,第一阶段搅拌反应时的温度为:1~20℃,搅拌反应时间为:1~3小时;
57.所述加完碳酸钾后,第二阶段搅拌反应时的温度为:80~100℃,搅拌反应时间为:8~16小时;
58.其中,对悬浊液进行降温、过滤、水洗时的温度均为:1~10℃;
59.洗涤滤饼时,洗涤至水层的ph值处于7~10则停止洗涤;
60.所述步骤3.3还包括:
61.所述溶剂三为:甲醇,或乙醇、四氢呋喃,1,4-二氧六环中的一种或多种混合物;所述溶剂三的用量为:滤饼(湿重)的3~5倍质量比;对所述滤饼进行加热搅拌萃取、和趁热过滤时的温度为:60~80℃;所述搅拌萃取的时间为:1~2小时。
62.所述步骤3.4包括:
63.结晶提纯时的溶剂四为:甲醇、乙醇、丙醇、异丙醇中的其中一种或多种混合物;
64.所述结晶提纯时的溶剂四用量为湿重的2-(4-硝基)苯基-5-氨基苯并咪唑粗品的4~6倍质量比。
65.9优选的,所述步骤4还包括:
66.步骤4.1,把2-(4-硝基)苯基-4-氨基苯并咪唑与溶剂五、加氢催化剂混合,放入高压反应釜;依次用氮气、氢气置换釜内气氛后,控制温度在30~100℃、氢气压力在0.3~1.5mpa持续加氢;直到取样用薄层色谱层析法进行分析原料均转化为2-(4-氨基)苯基-4-氨基苯并咪唑混合物为止;
67.步骤4.2,把步骤4.1中反应完毕后的2-(4-氨基)苯基-4-氨基苯并咪唑混合物,降温至常温并氮气置换后出料,过滤除去加氢催化剂;所得滤液浓缩掉溶剂五,即得2-(4-氨基)苯基-4-氨基苯并咪唑的粗品。
68.优选的,所述步骤4.1还包括:
69.所述溶剂五为:甲醇、乙醇、丙醇、异丙醇、丁醇、异丁醇、吡啶、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮中的一种或多种混合物;
70.所述溶剂五的用量为:2-(4-硝基)苯基-4-氨基苯并咪唑的4~20 倍质量比;
71.所述加氢催化剂为:用于硝基加氢的催化剂,催化剂包括并不限于为钯炭、铂碳、兰尼镍;
72.所述加氢催化剂的用量为:2-(4-硝基)苯基-4-氨基苯并咪唑的 0.5%~30%质量比;
73.所述加氢反应时的温度为:30~100℃;
74.所述加氢反应时的氢气压力为:0.3~1.5mpa;
75.本发明的工作原理和有益效果如下:
76.本发明所述的制备2-(4-氨基)苯基-4-氨基苯并咪唑的整条工艺路线为全新设计,在本发明之前未见其他文献的类似报道;
77.本发明使用4-(4-硝基-α-亚氨基)苯甲氨基苯甲酰胺来制备2
‑ꢀ
(4-硝基)苯基-5-氨基苯并咪唑;并在该反应中,创造性的使用同一种反应条件,不但使得原料上的苯甲酰胺基团转化为苯胺基团,而且同时使得原料上的亚氨基也发生关环反应,生成咪唑环(反应方程式如下);在本发明之前未见过其他文献的类似报道。
78.79.本方法所述的工艺路线,避开了现有其他同类用途工艺路线存在的多种问题,如使用昂贵、而且危险易爆的(同苯环)多硝基化合物;或多氨基反应的选择性差等缺陷。相对现有的、已实现工业化的其他工艺路线,本发明的整体收率接近、但原材料价格更为低廉;而且安全性更高、产生的有害污染物更少。
80.本发明的其它特征和优点将在随后的说明书中阐述,并且,部分地从说明书中变得显而易见,或者通过实施本发明而了解。本发明的目的和其他优点可通过在所写的说明书中所特别指出的结构来实现和获得。
81.下面通过实施例,对本发明的技术方案做进一步的详细描述。
具体实施方式
82.以下对本发明的优选实施例进行说明,应当理解,此处所描述的优选实施例仅用于说明和解释本发明,并不用于限定本发明。
83.本发明实施例提供了一种2-(4-氨基苯基)-5-氨基苯并咪唑的合成方法,包括:
84.步骤1,制备n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯;
85.步骤2,利用步骤1中获得的n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯制备4-(4-硝基-α-亚氨基)苯甲氨基苯甲酰胺;
86.步骤3,一锅法制备2-(4-硝基)苯基-5-氨基苯并咪唑;
87.步骤4,把2-(4-硝基)苯基-5-氨基苯并咪唑用加氢催化剂和氢气进行加氢反应,得到2-(4-氨基)苯基-4-氨基苯并咪唑。
88.本发明所述的制备2-(4-氨基)苯基-4-氨基苯并咪唑的整条工艺路线为全新设计,在本发明之前未见其他文献的类似报道;
89.本发明使用4-(4-硝基-α-亚氨基)苯甲氨基苯甲酰胺来制备2
‑ꢀ
(4-硝基)苯基-5-氨基苯并咪唑;并在该反应中,创造性的使用同一种反应条件,不但使得原料上的苯甲酰胺基团转化为苯胺基团,而且同时使得原料上的亚氨基也发生关环反应,生成咪唑环(反应方程式如下);在本发明之前未见过其他文献的类似报道。
[0090][0091]
本方法所述的工艺路线,避开了现有其他同类用途工艺路线存在的多种问题,如使用昂贵、而且危险易爆的(同苯环)多硝基化合物;或多氨基反应的选择性差等缺陷。相对现有的、已实现工业化的其他工艺路线,本发明的整体收率接近、但原材料价格更为低廉;而且安全性更高、产生的有害污染物更少。
[0092]
进一步的,所述步骤1包括:先使用对硝基苯甲酰氯与对氨基苯甲酸反应得到n-(4-硝基)苯甲酰基对氨基苯甲酸;再加入氯化亚砜反应得到n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯。
[0093]
所述步骤1还包括:
[0094]
步骤1.1,向溶剂一中加入对氨基苯甲酸;然后于0~30℃滴加对硝基苯甲酰氯溶液溶于溶剂一中;滴加完毕后,于0~30℃搅拌继续 2~10小时,生成n-(4-硝基)苯甲酰基
对氨基苯甲酸在溶剂中的悬浮物。
[0095]
步骤1.2,把步骤1.1所得的反应混合物升温至75~85℃,然后滴加氯化亚砜;滴加完毕后,继续保温75~85℃反应5~10小时;然后减压浓缩除去溶剂一和过量的氯化亚砜,即得n-(4-硝基-α-氯代) 苯亚甲基对氨基苯甲酰氯的粗品。
[0096]
其中,所述步骤1.1中还包括:
[0097]
所述溶剂一为:1,2-二氯乙烷、甲苯、二甲苯、氯苯中的一种,或它们的混合物;所述溶剂一的用量为:对氨基苯甲酸的10~20倍质量比;所述对硝基苯甲酰氯的用量为:对苯二胺的1.05~1.2倍当量;所述对硝基苯甲酰氯溶液为:对硝基苯甲酰氯与所述溶剂一配制而成,溶剂的配置用量为:在0~30℃把对硝基苯甲酰氯全部溶解;滴加对硝基苯甲酰氯溶液的滴加温度和反应温度为:0~30℃;滴加对硝基苯甲酰氯溶液完毕后的保温反应时间为:2~10小时;
[0098]
其中,所述步骤1.2中还包括:
[0099]
所述氯化亚砜用量为:对氨基苯甲酸的2.5~3.5倍当量;滴加氯化亚砜时和滴加后的保温反应温度为:75~85℃;滴加氯化亚砜后的保温反应时间为:5~10小时。
[0100]
所述步骤2还包括:
[0101]
把步骤1.2所得n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯粗品与溶剂二混合、溶清;把活性炭悬浮在水中,搅拌下控温1~30℃,先加入氨水;然后滴加并滴加前述配好的、n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯的溶液;滴加完毕后,继续保温1~30℃搅拌反应3~6小时,过滤;滤饼经水洗除去大部分的副产物氯化铵后,即得吸附在活性炭上的4-(4-硝基-α-亚氨基)苯甲氨基苯甲酰胺湿品。
[0102]
所述步骤2中还包括:
[0103]
所述溶剂二为:1,2-二氯乙烷、二氯甲烷、氯仿、甲苯中的一种或多种混合物;所述溶剂二的用量为:n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯粗品的4~10倍质量比;所述活性炭粒度为: 100~300目;所述活性炭用量为:n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯的5~10倍质量比;所述水的用量为:n-(4-硝基-α
‑ꢀ
氯代)苯亚甲基对氨基苯甲酰氯粗品的25~50倍质量比;所述氨水的用量为:n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯的2.5~4.0倍当量;其中,加入氨水、滴加n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯溶液时的温度为:1~30℃;所述n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯溶液完毕后的反应温度为:1~30℃,反应时间为: 3~6小时;所述过滤、用水洗涤滤饼时的时的温度为0℃~室温;洗涤滤饼时,洗涤至水层的ph值处于7~10。
[0104]
所述步骤3还包括:
[0105]
步骤3.1,把步骤2所得吸附在活性炭上的4-(4-硝基-α-亚氨基) 苯甲氨基苯甲酰胺湿品悬浮在水中,一边搅拌一边控制液温在 20~40℃,先滴加盐酸;再滴加次氯酸钠水溶液;加完后继续控制液温20~40℃搅拌1~3小时,获得悬浊液;
[0106]
步骤3.2,把步骤3.1所得的悬浊液搅拌下降温至1~20℃,然后分批加入碳酸钾;加完后,先继续控制液温1~20℃搅拌1~3小时;再升温至至80~100℃搅拌反应8~16小时;最后降温至5~10℃,过滤;所得滤饼用水洗涤后,即得吸附在活性炭上的2-(4-硝基)苯基-5
‑ꢀ
氨基苯并咪唑,其中,所述2-(4-硝基)苯基-5-氨基苯并咪唑为湿品;
[0107]
步骤3.3,向步骤3.2所得的(水洗后的)滤饼中,加入溶剂三,并加热至60~80℃搅
拌萃取1~2小时,趁热过滤;
[0108]
步骤3.4,把步骤3.3所得滤饼,再重复一遍步骤3.2所述操作进行萃取;然后合并两次萃取的滤液,减压浓缩除去溶剂三后,即得 2-(4-硝基)苯基-5-氨基苯并咪唑粗品;将2-(4-硝基)苯基-5-氨基苯并咪唑粗品用4~6倍质量的醇类溶剂四结晶提纯、再真空烘干,得 2-(4-硝基)苯基-5-氨基苯并咪唑;其中,两次萃取后的滤饼为活性炭,所述活性炭可回收。
[0109]
其中,步骤3.1还包括:
[0110]
所述水的用量为:活性炭及4-(4-硝基-α-亚氨基)苯甲氨基苯甲酰胺湿品的总质量的3~6倍;所述盐酸的用量为:2.1中所用对氨基苯甲酸的4.0~5.0倍当量;所述次氯酸钠的用量为:2.1中所用对氨基苯甲酸的2.5~3.5倍当量;加入盐酸、次氯酸钠时的温度,及加完后搅拌时的温度均为:20~40℃;所述加完盐酸、次氯酸钠后搅拌的时间均为1~3小时;
[0111]
所述步骤3.2还包括:
[0112]
所述碳酸钾的用量为:2.1中所用对氨基苯甲酸的2.5~3.5倍当量;所述加入碳酸钾时的温度为:1~20℃;所述加完碳酸钾后,第一阶段搅拌反应时的温度为:1~20℃,搅拌反应时间为:1~3小时;所述加完碳酸钾后,第二阶段搅拌反应时的温度为:80~100℃,搅拌反应时间为:8~16小时;其中,对悬浊液进行降温、过滤、水洗时的温度均为:1~10℃;洗涤滤饼时,洗涤至水层的ph值处于7~10则停止洗涤;
[0113]
所述步骤3.3还包括:
[0114]
所述溶剂三为:甲醇,或乙醇、四氢呋喃,1,4-二氧六环中的一种或多种混合物;所述溶剂三的用量为:滤饼(湿重)的3~5倍质量比;对所述滤饼进行加热搅拌萃取、和趁热过滤时的温度为:60~80℃;所述搅拌萃取的时间为:1~2小时。
[0115]
所述步骤3.4包括:
[0116]
结晶提纯时的溶剂四为:甲醇、乙醇、丙醇、异丙醇中的其中一种或多种混合物;所述结晶提纯时的溶剂四用量为湿重的2-(4-硝基) 苯基-5-氨基苯并咪唑粗品的4~6倍质量比。
[0117]
所述步骤4还包括:
[0118]
步骤4.1,把2-(4-硝基)苯基-4-氨基苯并咪唑与溶剂五、加氢催化剂混合,放入高压反应釜;依次用氮气、氢气置换釜内气氛后,控制温度在30~100℃、氢气压力在0.3~1.5mpa持续加氢;直到取样用薄层色谱层析法进行分析原料均转化为2-(4-氨基)苯基-4-氨基苯并咪唑混合物为止;
[0119]
进一步的,所述步骤4.1还包括:所述溶剂五为:甲醇、乙醇、丙醇、异丙醇、丁醇、异丁醇、吡啶、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮中的一种或多种混合物;所述溶剂五的用量为:2-(4-硝基)苯基-4-氨基苯并咪唑的4~20倍质量比;所述加氢催化剂为:用于硝基加氢的催化剂,催化剂包括并不限于为钯炭、铂碳、兰尼镍;所述加氢催化剂的用量为:2-(4-硝基)苯基-4-氨基苯并咪唑的0.5%~30%质量比;所述加氢反应时的温度为:30~100℃;所述加氢反应时的氢气压力为:0.3~1.5mpa。
[0120]
步骤4.2,把步骤4.1中反应完毕后的2-(4-氨基)苯基-4-氨基苯并咪唑混合物,降温至常温并氮气置换后出料,过滤除去加氢催化剂;所得滤液浓缩掉溶剂五,即得2-(4-氨
基)苯基-4-氨基苯并咪唑的粗品。
[0121]
具体的,参照以下具体步骤制备2-(4-氨基)苯基-4-氨基苯并咪唑:
[0122]
步骤1,一锅法制备n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯。
[0123]
步骤1包括:先使用对硝基苯甲酰氯与对氨基苯甲酸反应得到 n-(4-硝基)苯甲酰基对氨基苯甲酸;再加入氯化亚砜反应得到n
‑ꢀ
(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯。
[0124][0125]
步骤1的具体操作如下:
[0126]
步骤1.1、向溶剂中加入对氨基苯甲酸;然后于0~30℃滴加对硝基苯甲酰氯溶于溶剂的溶液。滴加完毕后,于0~30℃之间搅拌继续 2~10小时,生成n-(4-硝基)苯甲酰基对氨基苯甲酸在溶剂中的悬浮物。
[0127]
所述溶剂可以是1,2-二氯乙烷、甲苯、二甲苯、氯苯中的一种,或它们的混合物;
[0128]
所述溶剂的用量可以是对氨基苯甲酸的10~20倍质量比;
[0129]
所述的对硝基苯甲酰氯的用量,可以是对苯二胺的1.05~1.2倍当量;
[0130]
所述的对硝基苯甲酰氯溶液,可用对硝基苯甲酰氯与前述溶剂配制,溶剂用量以可以在0~30℃把对硝基苯甲酰氯全部溶解即可;
[0131]
所述的滴加和反应温度,都在0~30℃之间;
[0132]
所述的滴加完毕后的保温反应时间,在2~10小时之间。
[0133]
步骤1.2、把步骤1.1所得的反应混合物升温至75~85℃,然后滴加氯化亚砜;滴加完毕后,继续保温75~85℃反应5~10小时。然后减压浓缩除去溶剂和过量的氯化亚砜,即得n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯的粗品。
[0134]
所述的氯化亚砜用量,可用是对氨基苯甲酸的2.5~3.5倍当量;
[0135]
所述的滴加操作时间无严格要求,以控制反应放热、保证反应液温度在75~85℃之间为宜;
[0136]
所述的滴加和滴加后的保温反应温度,都在75~85℃之间;
[0137]
所述的滴加后的保温反应时间,在5~10小时之间。
[0138]
步骤2,制备4-(4-硝基-α-亚氨基)苯甲氨基苯甲酰胺。
[0139][0140]
步骤2的具体操作:
[0141]
把步骤1中所得n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯粗品与溶剂混合、溶清。把活性炭悬浮在水中,搅拌下控温1~30℃,先加入氨水;然后滴加并滴加前述配好的、n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯的溶液。滴加完毕后,继续保温1~30℃搅拌
反应3~6小时,过滤。滤饼经水洗除去大部分的副产物氯化铵后,即得吸附在活性炭上的4-(4-硝基-α-亚氨基)苯甲氨基苯甲酰胺湿品;无需干燥或提纯。
[0142]
所述溶剂可以是1,2-二氯乙烷、二氯甲烷、氯仿、甲苯中的一种,或它们的混合物;
[0143]
所述溶剂的用量可以是n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯粗品的4~10倍质量比;
[0144]
所述的活性炭粒度在100~300目之间;
[0145]
所述的活性炭用量可以是n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯的5~10倍质量比;
[0146]
所述的水的用量可以是n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯粗品的25~50倍质量比;
[0147]
所述氨水的用量,可以是n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯的2.5~4.0倍当量;
[0148]
所述加入氨水、滴加n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯溶液时的温度,都在1~30℃之间;
[0149]
所述的n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯溶液完毕后的反应温度在1~30℃之间,反应时间在3~6小时之间;
[0150]
所述过滤、用水洗涤滤饼时的时的温度,为0℃~室温之间;
[0151]
所述洗涤滤饼时,水的用量、温度、洗涤方式无固定要求,洗涤至水层的ph值处于7~10之间即可。
[0152]
步骤3,一锅法制备2-(4-硝基)苯基-5-氨基苯并咪唑。
[0153][0154]
步骤3的具体操作:
[0155]
步骤3.1、把步骤2中所得吸附在活性炭上的4-(4-硝基-α-亚氨基)苯甲氨基苯甲酰胺湿品悬浮在水中,一边搅拌一边控制液温在 20~40℃之间,先滴加盐酸;再滴加次氯酸钠水溶液;加完后继续控制液温20~40℃之间搅拌1~3小时。
[0156]
所述水的用量,可以是活性炭及4-(4-硝基-α-亚氨基)苯甲氨基苯甲酰胺湿品的总质量的3~6倍;
[0157]
所述盐酸的用量,可以是2.1中所用对氨基苯甲酸的4.0~5.0倍当量;
[0158]
所述次氯酸钠的用量,可以是2.1中所用对氨基苯甲酸的2.5~3.5 倍当量;
[0159]
所述加入盐酸、次氯酸钠时的温度,及加完后搅拌时的温度,都在20~40℃之间;
[0160]
所述加完盐酸、次氯酸钠后搅拌的时间,在1~3小时之间。
[0161]
步骤3.2、把步骤3.1所得的悬浊液搅拌下降温至1~20℃,然后分批加入碳酸钾。加完后,先继续控制液温1~20℃之间搅拌1~3小时;再升温至至80~100℃搅拌反应8~16小时。最后降温至5~10℃,过滤。所得滤饼用水洗涤后,即得吸附在活性炭上的2-(4-硝基)苯基-5-氨基苯并咪唑(湿品)。
[0162]
所述碳酸钾的用量,可以是2.1中所用对氨基苯甲酸的2.5~3.5 倍当量;
[0163]
所述加入碳酸钾时的温度,在1~20℃之间;
[0164]
所述加完碳酸钾后,第一阶段搅拌反应时的温度,在1~20℃之间,时间在1~3小时之间;
[0165]
所述加完碳酸钾后,第二阶段搅拌反应时的温度,在80~100℃之间,时间在8~16小时之间;
[0166]
所述降温、过滤、水洗时的温度,在1~10℃之间;
[0167]
所述洗涤滤饼时,水的用量、温度、洗涤方式无固定要求,洗涤至水层的ph值处于7~10之间即可。
[0168]
步骤3.3、向步骤3.2所得的(水洗后的)滤饼中,加入溶剂,并加热至60~80℃搅拌萃取1~2小时,趁热过滤。
[0169]
所述溶剂可以是甲醇,或乙醇、四氢呋喃,1,4-二氧六环的一种,或它们的混合物;
[0170]
所述溶剂的用量,可以是滤饼(湿重)的3~5倍质量比;
[0171]
所述加热搅拌萃取、和趁热过滤时的温度,在60~80℃之间;
[0172]
所述搅拌萃取的时间,可以是1~2小时。
[0173]
步骤3.4、把步骤3.3所得滤饼,再重复一遍步骤3.2所述的操作 (再萃取一次)。然后合并两次萃取的滤液,减压浓缩除去溶剂后,即得2-(4-硝基)苯基-5-氨基苯并咪唑的粗品。该粗品再用4~6倍质量的醇类溶剂结晶提纯、再真空烘干,既得2-(4-硝基)苯基-5-氨基苯并咪唑。经两次萃取后的滤饼为活性炭,回收。
[0174]
所述第二次萃取时的条件和操作,与步骤3.3完全相同。
[0175]
所述结晶提纯时的溶剂,可使用甲醇、乙醇、丙醇、异丙醇等,或是它们的混合物;
[0176]
所述结晶提纯时的溶剂用量,可以是2-(4-硝基)苯基-5-氨基苯并咪唑的粗品(湿重)的4~6倍质量比。
[0177]
步骤4,把2-(4-硝基)苯基-5-氨基苯并咪唑用加氢催化剂和氢气进行加氢反应,得到2-(4-氨基)苯基-4-氨基苯并咪唑。
[0178][0179]
步骤4的具体操作:
[0180]
步骤4.1、把2-(4-硝基)苯基-4-氨基苯并咪唑与溶剂、加氢催化剂混合,放入高压反应釜;依次用氮气、氢气置换釜内气氛后,控制温度在30~100℃之间、氢气压力在0.3~1.5mpa之间持续加氢;直到取样用薄层色谱层析法进行分析时,发现原料都转化为2-(4-氨基) 苯基-4-氨基苯并咪唑为止。
[0181]
所述溶剂可以是甲醇、乙醇、丙醇、异丙醇、丁醇、异丁醇、吡啶、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮中的一种,或它们的混合物;
[0182]
所述溶剂的用量,可以是2-(4-硝基)苯基-4-氨基苯并咪唑的 4~20倍质量比;
[0183]
所述加氢催化剂,可以是各种用于硝基加氢的常用催化剂,如钯炭、铂碳、兰尼镍,等等;
[0184]
所述加氢催化剂的用量,可以是2-(4-硝基)苯基-4-氨基苯并咪唑的0.5%~30%
质量比;
[0185]
所述加氢反应时的温度,可以在30~100℃之间;
[0186]
所述加氢反应时的氢气压力,可以在0.3~1.5mpa之间;
[0187]
所述加氢反应时的时间无固定要求,以原料都转化为2-(4-氨基) 苯基-4-氨基苯并咪唑(取样用薄层色谱层析法进行分析)为准。
[0188]
步骤4.2、把步骤4.1中反应完毕后的混合物,降温至常温并氮气置换后出料,过滤除去加氢催化剂;所得滤液浓缩掉溶剂,即得 2-(4-氨基)苯基-4-氨基苯并咪唑的粗品。
[0189]
本发明所述的制备2-(4-氨基)苯基-4-氨基苯并咪唑的整条工艺路线为全新设计,在本发明之前未见其他文献的类似报道;
[0190]
本发明使用4-(4-硝基-α-亚氨基)苯甲氨基苯甲酰胺来制备2
‑ꢀ
(4-硝基)苯基-5-氨基苯并咪唑;并在该反应中,创造性的使用同一种反应条件,不但使得原料上的苯甲酰胺基团转化为苯胺基团,而且同时使得原料上的亚氨基也发生关环反应,生成咪唑环(反应方程式如下);在本发明之前未见过其他文献的类似报道。
[0191][0192]
本方法所述的工艺路线,避开了现有其他同类用途工艺路线存在的多种问题,如使用昂贵、而且危险易爆的(同苯环)多硝基化合物;或多氨基反应的选择性差等缺陷。相对现有的、已实现工业化的其他工艺路线,本发明的整体收率接近、但原材料价格更为低廉;而且安全性更高、产生的有害污染物更少。
[0193]
实验例
[0194]
步骤1,一锅法制备n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯。
[0195][0196]
实验例1.1
[0197]
向1000ml反应瓶中,加入13.7g(0.1mol)的对氨基甲酸、274g的甲苯。搅拌下,保温20~30℃滴加19.5g(0.105mol)对硝基苯甲酰氯溶于100g甲苯的溶液。滴加完毕后,于20~30℃之间搅拌继续2小时。
[0198]
把反应液搅拌下升温至80~85℃,然后滴加41.7g(0.35mol)的氯化亚砜;滴加完毕后,继续保温80~85℃反应5小时。然后减压浓缩除去甲苯和过量的氯化亚砜,即得n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯的粗品约34.1g,收率≥100%。
[0199]
实验例2.1
[0200]
向500ml反应瓶中,加入13.7g(0.1mol)的对氨基甲酸、137g的 1,2-二氯乙烷。搅拌下,保温0~10℃滴加22.3g(0.12mol)对硝基苯甲酰氯溶于100g的1,2-二氯乙烷的溶液。滴加完毕后,于0~10℃之间搅拌继续10小时。
[0201]
把反应液搅拌下升温至75~80℃,然后滴加29.8g(0.25mol)的氯化亚砜;滴加完
毕后,继续保温75~80℃反应10小时。然后减压浓缩除去甲苯和过量的氯化亚砜,即得n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯的粗品约36.2g,收率≥100%。
[0202]
步骤2,制备4-(4-硝基-α-亚氨基)苯甲氨基苯甲酰胺。
[0203][0204]
实验例1.2
[0205]
先把实验例1.1所得的n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯(质量34.1g g,理论当量0.1mol)溶于171g的甲苯。再于2000ml 反应瓶中,加入852g的水和171g的活性炭(200~300目)。搅拌下,控温20~30℃加入约0.25mol的氨水;然后继续保温20~30℃,搅拌并滴加前述配好的n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯溶于甲苯的溶液。滴加完毕后,继续保温20~30℃反应3小时,然后室温下过滤。滤饼再室温下用水冲洗,至水层的ph值达到7~10之间。洗涤完毕后的滤饼为吸附在活性炭上的4-(4-硝基-α-亚氨基)苯甲氨基苯甲酰胺,湿重约281g,无需干燥。
[0206]
实验例2.2
[0207]
先把实验例2.1所得的n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯(质量36.2g g,理论当量0.1mol)溶于362g的1,2-二氯乙烷。再于3000ml反应瓶中,加入1810g的水和362g的活性炭(100~200 目)。搅拌下,控温1~10℃加入约0.4mol的氨水;然后继续保温1~10℃,搅拌并滴加前述配好的n-(4-硝基-α-氯代)苯亚甲基对氨基苯甲酰氯溶于1,2-二氯乙烷的溶液。滴加完毕后,继续保温1~10℃搅拌反应 6小时,然后于0~10℃,过滤。滤饼再用0~10℃的水冲洗,至水层的ph值达到7~10之间。洗涤完毕后的滤饼为吸附在活性炭上的4
‑ꢀ
(4-硝基-α-亚氨基)苯甲氨基苯甲酰胺,湿重约527g,无需干燥。
[0208]
步骤3,一锅法制备2-(4-硝基)苯基-5-氨基苯并咪唑。
[0209][0210]
实验例1.3
[0211]
步骤1.3.1、把实验例1.2所得的吸附在活性炭上的4-(4-硝基
‑ꢀ
α-亚氨基)苯甲氨基苯甲酰胺(湿重总计约281g,理论当量0.1mol) 加入到3000ml反应瓶中,再加入1686g的水。搅拌下控制液温 30~40℃,先加入0.4mol的盐酸;再滴加总计0.25mol的次氯酸钠水溶液。加完后继续保温30~40℃搅拌1小时,获得反应液。
[0212]
步骤1.3.2、把1.3.1所得反应液降温至10~20℃,搅拌下分批加入总计34.5g(0.25mol)的碳酸钾。加完后,先继续保温10~20℃搅拌1小时;然后升温至80~90℃,搅拌反应16小时。之后把反应液降温至1~10℃,过滤;所得滤饼用1~10℃的水冲洗,至水层的ph值在 7~10之间后,得滤饼(湿品)约272g。
[0213]
步骤1.3.3、向步骤1.3.2所得的滤饼中,加入1360g的四氢呋喃,并加热搅拌至60
~65℃回流1小时。趁热过滤;滤饼再用1360g的四氢呋喃加热回流萃取、过滤一次。两次的滤液合并后,减压浓缩除去四氢呋喃,剩余物即为2-(4-硝基)苯基-5-氨基苯并咪唑的粗品,约 23.6g。二次萃取后的滤饼为活性炭,回收。
[0214]
步骤1.3.4、把1.3.3所得2-(4-硝基)苯基-5-氨基苯并咪唑的粗品,用141.6g的乙醇结晶一次、真空烘干后,得2-(4-硝基)苯基-5
‑ꢀ
氨基苯并咪唑约20.8g,液相色谱纯度≥99%,收率82%。
[0215]
实验例2.3
[0216]
步骤2.3.1、把实验例2.2所得的吸附在活性炭上的4-(4-硝基
‑ꢀ
α-亚氨基)苯甲氨基苯甲酰胺(湿重总计约527g,理论当量0.1mol) 加入到3000ml反应瓶中,再加入1581g的水。搅拌下控制液温 20~30℃,先加入0.5mol的盐酸;再滴加总计0.35mol的次氯酸钠水溶液。加完后继续保温20~30℃搅拌3小时,获得反应液。
[0217]
步骤2.3.2、把步骤2.3.1所得反应液降温至1~10℃,搅拌下分批加入总计48.3g(0.35mol)的碳酸钾。加完后,先继续保温1~10搅拌3 小时;然后升温至90~100℃,搅拌反应8小时。之后把反应液降温至1~10℃,过滤;所得滤饼用1~10℃的水冲洗,至水层的ph值在 7~10之间后,得滤饼(湿品)约505g。
[0218]
步骤2.3.3、向步骤2.3.2所得滤饼中,加入1515g的乙醇,并加热搅拌至70~80℃回流1小时。趁热过滤;滤饼再用1515g的乙醇加热回流萃取、过滤一次。两次的滤液合并后,减压浓缩除去乙醇,剩余物即为2-(4-硝基)苯基-5-氨基苯并咪唑的粗品,约24.2g。二次萃取后的滤饼为活性炭,回收。
[0219]
步骤2.3.4、把步骤2.3.3所得2-(4-硝基)苯基-5-氨基苯并咪唑的粗品,用96.8g的甲醇结晶一次、真空烘干后,得2-(4-硝基)苯基-5-氨基苯并咪唑约21.3g,液相色谱纯度≥99%,收率84%。
[0220]
步骤4,合成2-(4-氨基)苯基-4-氨基苯并咪唑。
[0221][0222]
实验例1.4
[0223]
向高压反应釜中,加入20.8g的2-(4-硝基)苯基-4-氨基苯并咪唑、84.4g的n,n-二甲基甲酰胺、和6.33g的兰尼镍。依次用氮气置换、氢气置换釜内气氛后,搅拌下慢慢升温至90~100℃,氢气加压至1.4~1.5mpa,持续反应;并间或取出反应液,以薄板层析法分析原料转化为目标产物的转化率。约5~8小时后转化完全;把反应液降温至接近室温。再氮气置换后,取出反应混合物,并过滤除去兰尼镍;所得滤液经减压浓缩除去大部分溶剂后,得到棕黑色的浓缩剩余物。用乙酸乙酯洗涤一次、真空干燥后,得17.1g的2-(4-硝基)苯基-4
‑ꢀ
氨基苯并咪唑,收率98%,液相色谱纯度≥98%。
[0224]
实验例2.4
[0225]
向高压反应釜中,加入21.4g的2-(4-硝基)苯基-4-氨基苯并咪唑、432g的乙醇、和0.11g的钯炭。依次用氮气置换、氢气置换釜内气氛后,搅拌下控温30~40℃,氢气加压至0.3~0.4mpa,持续反应;并间或取出反应液,以薄板层析法分析原料转化为目标产物的转
化率。约24~36小时后转化完全;把反应液降温至接近室温。再氮气置换后,取出反应混合物,并过滤除去钯炭;所得滤液经减压浓缩除去大部分溶剂后,得到棕黑色的2-(4-氨基)苯基-4-氨基苯并咪唑粗品。用乙酸乙酯洗涤一次、真空干燥后,得18.6g的2-(4-硝基)苯基-4-氨基苯并咪唑,收率99%,液相色谱纯度≥97%。
[0226]
综上,本发明提供的2-(4-硝基)苯基-4-氨基苯并咪唑合成工艺路线,避开了现有其他同类用途工艺路线存在的多种问题,如使用昂贵、而且危险易爆的(同苯环)多硝基化合物;或多氨基反应的选择性差等缺陷。相对现有的、已实现工业化的其他工艺路线,本发明的整体收率接近、但原材料价格更为低廉;而且安全性更高、产生的有害污染物更少,因此,更利于大规模生产。
[0227]
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。