一种17
α-孕烯醇酮的制备方法
技术领域
1.本发明涉及医药领域,具体涉及一种别孕烷醇酮有关物质的制备方法,特别是涉及17α-孕烯醇酮的制备方法。
背景技术:
2.别孕烷醇酮(brexanolone),化学名为:(3α,5α)-3-羟基-孕甾-20-酮,结构式如下,是一种神经活性类固醇物质。2019年,sage therapeutics公司在美国上市了别孕烷醇酮注射液,商品名为:zulresso,上市适应症为:成人产后抑郁症(ppd)。
[0003][0004]
别孕烷醇酮及其衍生物是近年来研究的热点,有多家医药公司正在开发别孕烷醇酮类衍生物。现有技术中别孕烷醇酮多是由孕烯醇酮(如下式1化合物)制备得到,在制备过程中孕烯醇酮会产生与其结构类似的有关物质,所述有关物质中主要有17α-孕烯醇酮。
[0005][0006]
任何影响药物纯度的物质统称为杂质,药物杂质一般包括:有机杂质、无机杂质及残留溶剂。有机杂质主要指由生产工艺引入或物质降解而产生的杂质,有机杂质的化学结构一般与活性成分类似或具渊源关系,故通常又可称之为有关物质。“有关物质”是指“在既定工艺进行生产和正常贮藏过程中可能含有或产生并需要控制的杂质,改变生产工艺时需另考虑增修订有关项目”。
[0007]
出于人用药安全考虑,在药物活性成分的产品商业化之前,国内和国际药品监管机构都会建立很低的杂质质控限度。通常已知杂质的质控限度为0.15%,但未知杂质的质控限度通常会小于0.10%。杂质的研究是药品研发的一项重要内容,它包括选择合适的分析方法,准确地分辨与测定杂质的含量并综合药学、毒理、临床研究的结果确定杂质的合理限度。
[0008]
在医药质量分析技术领域内,活性药物成分及杂质中的化学衍生物、合成副产物以及降解产物可采用光谱、色谱或其他物理方法鉴别或定量。在对化合物中的杂质进行分析前,需要采用纯度较高且与上述杂质相同或相近结构的物质作为参比标示物,并将参比标示物在色谱图中的相对位置视为杂质在色谱图中的相对位置,并以此对待检化合物的杂
质检测进行指导。显然,参比标示物的选择和制备,对活性药物成分中杂质含量检测的科学性和准确性有着直接的影响。
技术实现要素:
[0009]
本发明目的在于提供了一种高纯度的别孕烷醇酮有关物质化合物2(即17α-孕烯醇酮)的制备方法,该制备方法制备工艺简单,无需手性柱进行拆分,制备得到的化合物2纯度高,且后处理方便,更适宜于工业化大生产。17α-孕烯醇酮的化学结构式如下:
[0010][0011]
为了实现上述发明目的,本发明提供了以下技术方案:
[0012]
第一方面,本发明提供了一种17α-孕烯醇酮的制备方法,所述方法包含以下步骤:
[0013]
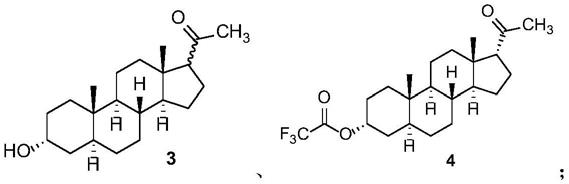
(1)将化合物1与溶剂混合,在碱存在的条件下反应,反应完全后,调节ph至6~9,过滤得式3混合物,
[0014][0015]
(2)在足以产生化合物4的反应条件下,将所得的式3混合物与三氟乙酸酐反应,得到化合物4,
[0016][0017]
(3)将化合物4与溶剂混合,在催化剂存在的条件下,制备得到17α-孕烯醇酮。
[0018]
在上述17α-孕烯醇酮制备方法的一些实施方案中,其中,所述式3混合物指的是由化合物1与化合物2组成的混合物。
[0019]
在上述17α-孕烯醇酮制备方法的一些实施方案中,步骤(1)中,其中,所述碱为无机碱。在一些实施方案中,所述无机碱选自碳酸钾、碳酸钠、碳酸铯、氢氧化钾、氢氧化钠、甲醇钠、乙醇钠、磷酸钾、磷酸钠中的一种。在一些实施方案中,所述无机碱为氢氧化钾、氢氧化钠、甲醇钠中的一种。在某些实施方案中,所述无机碱的用量为1.0-8.0摩尔当量。在某些具体的实施方案中,所述无机碱的用量为2.0-4.0摩尔当量。
[0020]
在上述17α-孕烯醇酮制备方法的一些实施方案中,步骤(1)中,其中,所述溶剂选自甲醇、乙醇、四氢呋喃中的一种;所述步骤(1)反应可以在加热的条件下进行。在一些实施方案中,步骤(1)中,其中,所述溶剂为甲醇;所述步骤(1)反应可以在加热至60-80℃条件下反应,例如,可以加热至在60-70℃、70-80℃条件下反应。
[0021]
在上述17α-孕烯醇酮制备方法的一些实施方案中,步骤(2)中,其中,所述反应条件包含缚酸剂和催化剂;所述缚酸剂选自三乙胺、吡啶中的一种,所述缚酸剂的用量为1.05-2.0摩尔当量;所述催化剂为4-二甲氨基吡啶,所述催化剂的用量为0.1-1.0摩尔当量。
[0022]
在上述17α-孕烯醇酮制备方法的一些实施方案中,步骤(2)中,其中,三氟乙酸酐的用量为1.0-2.0摩尔当量;在本发明的一些方案中,三氟乙酸酐的用量为1.1-1.5摩尔当量。
[0023]
在上述17α-孕烯醇酮制备方法的一些实施方案中,步骤(2)中,其中,所述溶剂选自二氯甲烷和四氢呋喃中的一种;在本发明的一些方案中,所述溶剂选自二氯甲烷。
[0024]
在上述17α-孕烯醇酮制备方法的一些实施方案中,步骤(3)中,其中,所述催化剂为醋酸钠,所述醋酸钠的用量为1.0-1.5摩尔当量;所述溶剂为甲醇、乙醇中的一种或其组合。
[0025]
化合物2的制备方法的具体反应路线如下:
[0026][0027]
第二方面,本发明还提供了一种17α-孕烯醇酮的制备方法,所述方法包含以下步骤:
[0028]
(1)在足以产生化合物4的反应条件下,将所得的式3混合物与三氟乙酸酐反应,得到化合物4,
[0029][0030]
(2)将化合物4与溶剂混合,在催化剂存在的条件下,制备得到17α-孕烯醇酮。
[0031]
在上述17α-孕烯醇酮制备方法的一些实施方案中,其中,所述式3混合物指的是由化合物1与化合物2组成的混合物。
[0032]
在上述17α-孕烯醇酮制备方法的一些实施方案中,步骤(1)中,其中,所述反应条件包含缚酸剂和催化剂;所述缚酸剂选自三乙胺、吡啶中的一种,所述缚酸剂的用量为1.05-2.0摩尔当量;所述催化剂为4-二甲氨基吡啶,所述催化剂的用量为0.1-1.0摩尔当量。
[0033]
在上述17α-孕烯醇酮制备方法的一些实施方案中,步骤(1)中,其中,三氟乙酸酐的用量为1.0-2.0摩尔当量;在本发明的一些方案中,三氟乙酸酐的用量为1.1-1.5摩尔当量。
[0034]
在上述17α-孕烯醇酮制备方法的一些实施方案中,步骤(1)中,其中,所述溶剂选自二氯甲烷和四氢呋喃中的一种;在本发明的一些方案中,所述溶剂选自二氯甲烷。
[0035]
在上述17α-孕烯醇酮制备方法的一些实施方案中,步骤(2)中,其中,所述催化剂为醋酸钠,所述醋酸钠的用量为1.0-1.5摩尔当量;所述溶剂为甲醇、乙醇中的一种或其组合。
[0036]
化合物2的制备方法的具体反应路线如下:
[0037][0038]
有益效果:
[0039]
本发明提供了一种高纯度的17α-孕烯醇酮的制备方法,该方法制备得到的17α-孕烯醇酮纯度高,制备方法简单,适于工业化大生产。17α-孕烯醇酮(即化合物2)与孕烯醇酮(即化合物1)为立体异构体,物理化学性质极其近似,利用常规的理化方法难以分离,大多通过手性高效液相方法进行分离、纯化。通过本发明提供的17α-孕烯醇酮制备方法制备得到的17α-孕烯醇酮纯度大于99.80%,且不需要手性高效液相方法等特殊的拆分方法,制备方法简单,适用于工业化生产。
[0040]
本发明方法制备得到的高纯度17α-孕烯醇酮能够在别孕烷酮的杂质检测时作为参比标示物,能够有效提高别孕烷酮、别孕烷酮类衍生物及其制剂中杂质检测的科学性和
准确性,能有效地、方便地监控别孕烷酮、别孕烷酮类衍生物及其制剂的杂质含量,保证别孕烷酮、别孕烷酮类衍生物及其制剂的安全性和有效性。
具体实施方式
[0041]
下文将结合具体实施例对本发明的通式化合物及其制备方法和应用做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
[0042]
定义和说明
[0043]
除非另有说明,本文所用的下列术语和短语旨在含有下列含义。一个特定的短语或术语在没有特别定义的情况下不应该被认为是不确定的或不清楚的,而应该按照普通的含义去理解。当本文出现商品名时,旨在指代其对应的商品或其活性成分。
[0044]
本发明中,术语“酸”、“过氧化物”、“碱”,可以选择直接加入反应体系中,也可以根据本领域技术人员的操作习惯稀释或配制成溶液后加入,以有效成分物质的量相同为准;术语“金属氢氧化物”如果可以含有结晶水,在反应过程中可以添加不含结晶水或含有结晶水的形式进行反应,以物质的量相同为准。
[0045]
本发明中,术语“碱”旨在表示这样的化学物质,其是质子受体。用于本发明的合适的碱为无机碱。无机碱的实例包括但不限于氢氧化钾(koh)、碳酸钾(k2co3)、碳酸钠(na2co3)、碳酸铯(cs2co3)、氢化钠(nah)、磷酸钾(k3po4)、磷酸钠(na3po4)等。
[0046]
术语“反应条件”是指表示化学反应进行的物理和/或环境条件,反应条件包括但不限于下面的一种或多种:反应温度、溶剂、ph值、压力、反应时间、反应物的摩尔比(以摩尔当量表示)、酸或碱、是否有催化剂的存在及催化剂的种类等。反应条件可以在其中使用所述条件的具体化学反应之后命名,例如偶合条件、氢化条件、酰基化条件、还原条件、氘代条件等。
[0047]
本发明中,“别孕烷酮”、“别孕烷醇酮”可以替换使用,均是指具有如下式结构的化合物:
[0048]
本发明中,“17α-孕烯醇酮”、“化合物2”可以替换使用,均是指具有如下式2结构的化合物:
[0049]
本发明提供的式2化合物制备方法的反应路线,具体如下:
[0050][0051]
本发明的发明人在制备化合物2的过程中发现,得到的混合物3为化合物1(即孕烯醇酮)与化合物2(即17α-孕烯醇酮)的混合物。化合物2与化合物1为立体异构体,物理化学性质极其近似,发明人尝试了多种常规的理化方法均难以将化合物2与化合物1相分离,最终发现手性色谱柱高效液相方法能够从混合物3中分离、纯化得到化合物2,但手性色谱柱高效液相方法难以应用于工业化生产。
[0052]
本发明的发明人最终发现,将混合物3与三氟乙酸酐反应,能够有效的分离得到化合物4,然后在不同的反应条件下尝试对化合物4进行水解制备成化合物2。发明人出人意料的发现当使用醋酸钠作为化合物4水解反应的催化剂时,能得到高纯度的化合物2,基本不会产生化合物1,本发明提供的化合物2制备方法制备得到的化合物2纯度大于99.80%,且基本不含有化合物1。
[0053]
本发明的中间体化合物可以通过本领域技术人员所熟知的多种合成方法来制备,包括下面列举的具体实施方式、其与其他化学合成方法的结合所形成的实施方式以及本领域技术人员所熟知的等同替换方式,优选的实施方式包括但不限于本发明的实施例。
[0054]
本发明具体实施方式的化学反应是在合适的溶剂中完成的,所述的溶剂须适合于本发明的化学变化及其所需的试剂和物料。为了获得本发明的化合物,有时需要本领域技术人员在已有实施方式的基础上对合成步骤或者反应流程进行修改或选择。
[0055]
下面会通过实施例具体描述本发明,这些实施例并不意味着对本发明的任何限制。
[0056]
下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另有说明,否则百分比和份数按重量计算,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
[0057]
测试方法:
[0058]
高效液相色谱(hplc)分析方法:
[0059]
所用仪器为:高效液相色谱仪agilent 1260;
[0060]
色谱柱为:phenomenex-ace excel 3 c18-ar 4.6mm
×
150mm;
[0061]
测定条件如下:
[0062]
进样体积:10μl;
[0063]
流速:1.0ml/min;
[0064]
检测波长:200nm;
[0065]
样品浓度:2.0mg/ml;
[0066]
稀释液:甲醇;
[0067]
柱温:35℃;
[0068]
流动相a:水;
[0069]
流动相b:乙腈;
[0070]
洗脱梯度如下表1:
[0071]
表1
[0072]
时间(min)%a%b06040202080255953559535.16040456040
[0073]
混合物3的制备
[0074][0075]
实施例1、混合物3的制备
[0076]
向10.0l三口圆底烧瓶中先加入141.80g koh(2528mmol,2.0eq),加入甲醇8000ml搅拌溶解,加入400.00g的化合物1(1264mmol,1.0eq)加热回流24小时,体系逐渐溶清,反应完将体系转移至25l容器中,搅拌下滴加2000ml水,析出固体,过滤,用甲醇/水混合溶剂(4v:1v)400ml洗涤,第一次滤饼抽干后得到固体,滤液浓缩至3l左右时,有大量固体析出,过滤得到第二次固体干燥得到36.30g。第一次滤饼重复koh异构化过程,经过相同处理,干燥后得到32.30g固体。两者合并得到混合物3(68.60g),混合物3为含化合物1与化合物2的混合物,经hplc测定,化合物2含量约为37.36%。
[0077]
实施例2、混合物3的制备
[0078]
向2.0l三口圆底烧瓶中先加入16.00g naoh(400mmol,4.0eq),加入甲醇634ml搅拌溶解,加入31.70g的化合物1(100mmol,1.0eq)加热回流4小时,反应完将体系转移至5l烧
杯,搅拌下滴加159ml水,析出固体,过滤,用甲醇/水混合溶剂(4v:1v)32ml洗涤,第一次滤饼抽干得到,滤液浓缩至240ml左右时,有固体析出,过滤得到第二次固体干燥得到2.88g。第一次滤饼重复naoh异构化过程,经过相同处理,干燥后得到2.51g固体。两者合并得到混合物3(5.39g),混合物3为含化合物1与化合物2的混合物,经hplc测定,化合物2含量约为36.82%。
[0079]
实施例3、混合物3的制备
[0080]
向2.0l三口圆底烧瓶中先加入6.40g甲醇钠(118.5mmol,1.0eq),加入甲醇750ml搅拌溶解,加入37.50g的化合物1(118.5mmol,1.00eq)加热回流4小时,反应完将体系转移至5l烧杯,搅拌下滴加188ml水,析出固体,过滤,用甲醇/水混合溶剂(4v:1v)38ml洗涤,第一次滤饼抽干后干燥,滤液浓缩至280ml左右时固体析出,过滤得到第二次固体干燥得到3.40g。第一次滤饼重复甲醇钠异构化过程,经过相同处理,干燥后得到3.32g固体。两者合并得到混合物3(6.72g),混合物3为含化合物1与化合物2的混合物,经hplc测定,化合物2含量约为37.15%。
[0081]
实施例4、混合物3的制备
[0082]
向2.0l三口圆底烧瓶中先加入84.80g碳酸钠(800mmol,8.0eq),加入甲醇634ml搅拌溶解,加入31.70g的化合物1(100mmol,1.0eq)加热回流4小时,反应完将体系转移至5l烧杯,搅拌下滴加159ml水,析出固体,过滤,用甲醇/水混合溶剂(4v:1v)32ml洗涤,第一次滤饼抽干得到,滤液浓缩至240ml左右时,有固体析出,过滤得到第二次固体干燥得到2.88g。第一次滤饼重复碳酸钠异构化过程,经过相同处理,干燥后得到2.51g固体。两者合并得到混合物3(5.39g),混合物3为含化合物1与化合物2的混合物,经hplc测定,化合物2含量约为36.95%。
[0083]
化合物4的制备
[0084][0085]
实施例5、化合物4的制备
[0086]
向250ml三口圆底烧瓶中加入6.55g混合物3(根据权利要求1方法制得,20.7mmol,1.0eq),66ml二氯甲烷,2.19g三乙胺(21.7mmol,1.05eq),2.53g dmap(20.7mmol,1.0eq),氮气保护冰水浴冷却,滴加4.56g三氟乙酸酐(21.7mmol,1.05eq),约15分钟滴加完成,搅拌1h左右,反应结束,33ml冰水淬灭反应后,分液,有机相用33ml纯水洗涤,有机相经过无水硫酸钠干燥后,柱层析分离(v石油醚(60-90):v乙酸乙酯=10:1),收集极性稍小点、减压蒸去溶剂,用15~20ml庚烷重结晶,过滤、真空干燥得到化合物4(2.67g),纯度为99.63%。
[0087]1h-nmr(500mhz,cdcl3)δδ5.40
–
5.29(m,1h),4.63-4.59(m,1h),2.91-2.83(m,1h),2.42-2.31(m,1h),2.29
–
2.20(m,1h),2.13(s,3h),2.04
–
1.87(m,2h),1.86-1.39(m,10h),1.30
–
1.13(m,3h),1.09
–
1.01(m,1h),1.00(s,3h),0.95
–
0.86(m,4h).ms m/z:435.21[m+na]
+
.
[0088]
实施例6、化合物4的制备
[0089]
向1l的三口瓶中加入39.00g混合物3(实施例1方法制得,122.4mmol)、390ml二氯甲烷,搅拌溶解。氮气保护下加入17.34g三乙胺(171.36mmol,1.40eq)和1.49g dmap(12.2mmol,0.10eq)。将三口瓶置于冰水浴中,缓慢滴加入38.56g三氟乙酸酐(183.6mmol,1.50eq),继续搅拌反应至反应完全。反应完全后,将反应液倒入200ml的冰水中淬灭反应,搅拌10min后分液,有机相用纯水洗涤2次(200ml*2)。有机相减压除去溶剂,得到固体物60.0g,柱层析分离(v石油醚(60-90):v乙酸乙酯=100:1),得到的化合物用120ml庚烷结晶,冷却至0~10℃,过滤,得到化合物4(10.98g),纯度为99.74%。
[0090]
实施例7、化合物4的制备
[0091]
向250ml的三口瓶中加入4.12g混合物3(实施例1方法制得,10.0mmol,1eq)、42ml二氯甲烷,搅拌溶解。氮气保护下加入2.02g三乙胺(20.0mmol,2.00eq)和0.244g dmap(2.0mmol,0.20eq)。将三口瓶置于冰水浴中,缓慢滴加入4.20g三氟乙酸酐(20.0mmol,2.00eq),继续搅拌反应至反应完全。反应完全后,将反应液倒入20ml的冰水中淬灭反应,搅拌10分钟后分液,有机相用纯水洗涤2次(20ml*2)。有机相减压除去溶剂,得到固体物60.0g,柱层析分离(v石油醚(60-90):v乙酸乙酯=100:1),得到的化合物用12ml庚烷结晶,冷却至-10~0℃,过滤,得到化合物4(1.32g),纯度为99.52%。
[0092]
化合物2的制备:
[0093][0094]
实施例8、化合物2的制备
[0095]
向100ml的单口瓶中加入1.67g的化合物4(实施例6方法制得,4.05mmol,1.0eq),0.33g无水乙酸钠(4.05mmol,1.0eq),甲醇15ml,加热回流。搅拌反应2~4h,tlc检测反应显示反应完全。降温至室温,瓶壁上析出少部分固体。滴加7.5ml的纯水。析出固体。过滤,滤饼用甲醇:水=1:1的混合溶剂洗涤(5ml*2)。滤饼抽干,于45度下真空干燥得化合物2(1.15g)收率90.0%,hplc纯度99.87%。
[0096]1h-nmr(500mhz,cdcl3)δ5.40
–
5.29(m,1h),3.58-3.48(m,1h),2.85-2.76(m,1h),2.33-2.27(m,1h),2.26
–
2.18(m,1h),2.13(s,3h),2.04
–
1.96(m,1h),1.95
–
1.87(m,1h),1.86
–
1.68(m,5h),1.67
–
1.39(m,6h),1.30
–
1.13(m,3h),1.09
–
1.01(m,1h),1.00(s,3h),0.95
–
0.86(m,4h).
13
c nmr(500mhz,cdcl3)δ212.81,140.43,121.64,71.62,61.29,50.53,49.50,45.48,42.21,37.23,36.46,35.09,32.82,32.03,32.01,31.27,26.03,24.35,21.02,20.63,19.36.ms m/z:317.28[m+h]
+
,633.60[2m+h]
+
.
[0097]
实施例9、化合物2的制备
[0098]
向100ml的单口瓶中加入4.12g化合物4(实施例6方法制得,10.0mmol)、25ml etoh、0.98g无水乙酸钠(12.0mmol,1.20eq),加毕,加热、搅拌回流反应至反应完全。反应液冷却至室温,滴加20.8ml纯水,搅拌、析出固体,过滤,滤饼用乙醇/水混合溶剂洗涤(v
etoh
:v
h2o
=1:1),干燥后得到化合物2纯品(2.95g收率93.3%),纯度为99.92%。
[0099]
实施例10、化合物2的制备
[0100]
向100ml的单口瓶中加入4.12g化合物4(实施例6方法制得,10.0mmol)、24.9ml meoh、1.23g无水乙酸钠(15.0mmol,1.50eq),加毕,加热、搅拌回流反应至反应完全。反应液冷却至室温,滴加20.8ml纯水,搅拌、析出固体,过滤,滤饼用甲醇/水混合溶剂洗涤(v
meoh
:v
h2o
=1:1),干燥后得到化合物2纯品(2.94g收率93.0%),纯度为99.91%。
[0101]
实施例11、不同碱作为水解条件的对比实验
[0102]
按实施例9的制备方法,分别以1.20eq的无机碱(分别为乙酸钠、苯甲酸钠、碳酸氢钠、氢氧化钠)作为水解反应催化剂,测得各无机碱对应的化合物2的收率和纯度,具体结果见下表2。
[0103]
具体反应步骤为:向100ml的单口瓶中加入4.12g化合物4(实施例6方法制得,10.0mmol)、25ml etoh及1.20eq的无机碱(乙酸钠、苯甲酸钠、碳酸氢钠、氢氧化钠),加毕,加热、搅拌回流反应至反应完全。反应液冷却至室温,滴加20.8ml纯水,搅拌、析出固体,过滤,滤饼用乙醇/水混合溶剂洗涤(v
etoh
:v
h2o
=1:1),干燥后得到化合物2,称重、计算收率,hplc检测纯度,具体结果见下表2。
[0104]
表2、不同无机碱催化水解实验的比较
[0105][0106]
通过上表2中的数据可以看出,以乙酸钠作为水解催化剂时,所得到的化合物2纯度高、收率好,适用于工业生产。高纯度的化合物2的制备,能极大的促进别孕烷酮、别孕烷酮衍生物及药品制剂的质量控制,确保药品的安全性。
[0107]
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。