1.本发明属于食品生物技术领域,具体涉及一种植物乳酸菌发酵上清中提取吲哚-3-乳酸的方法。
背景技术:
2.吲哚-3-乳酸(indole-3-lactic acid,ila),是一种带有吲哚环的色氨酸代谢产物,分子量为205da,分子式为c
11h11
no3,具有式i所示的结构。乳酸菌如长双歧杆菌、婴儿双歧杆菌、短双歧杆菌、唾液乳杆菌、植物乳杆菌、罗伊氏乳杆菌等可代谢产生吲哚-3-乳酸。与其他菌株相比,吲哚-3-乳酸是双歧杆菌产生的唯一色氨酸代谢物,从人类婴儿肠道分离的双歧杆菌菌株中发现的吲哚-3-乳酸含量相对较高,为22.17~33.12μg/ml。而植物乳杆菌菌株代谢产生吲哚-3-乳酸的菌株仅有植物乳杆菌um55、植物乳杆菌dy-1、植物乳杆菌f51等,含量为4.30~30.70μg/ml。
[0003][0004]
吲哚-3-乳酸(indole-3-lactic acid,ila)
[0005]
据报道,吲哚-3-乳酸具有抗氧化活性,参与诱导免疫调节,抑制内源性蛋白结合,作为酒精性肝病、胃癌诊断的保守生物标志物,对糖尿病、炎症性肠病和其他一些代谢疾病有效等功能作用。因此,吲哚-3-乳酸在医药、食品、保健等领域将有广泛应用。
[0006]
目前,从乳酸菌发酵上清液中分离吲哚-3-乳酸,主要采用超滤离心法。超滤离心截留的分子大小具有一定的范围,相近分子量的物质均被离心截留,且过滤器机械性能的不同,易导致分馏的吲哚-3-乳酸含量及纯度存在差异。因此,筛选高产吲哚-3-乳酸的乳酸菌,并优化分离纯化的方法对乳酸菌源吲哚-3-乳酸的提取意义十分重大。
[0007]
2008100625229的发明《lactobacillus plantarum zj316、产生的抗菌肽及其制备与应用》提供了一株从婴儿粪便中分离到的植物乳杆菌——lactobacillus plantarum zj316,保藏编号cctcc no:m 208077,所得的抗菌肽具有广谱的抑菌作用。
[0008]
201911341978.3的发明《直投式乳酸菌发酵剂和制备方法》提供了一直投式乳酸菌发酵剂和制备方法,其中所述直投式乳酸菌发酵剂,应用乳酸菌中的植物乳杆菌zj316制备,其中所述植物乳杆菌的保藏编号cctcc no:m 208077。
技术实现要素:
[0009]
本发明要解决的技术问题是提供一种从植物乳酸菌发酵上清中提取吲哚-3-乳酸的方法,并测定该吲哚-3-乳酸的抗菌活性。
[0010]
为解决上述技术问题,本发明提供一种从植物乳酸菌发酵上清中纯化吲哚-3-乳
酸的方法,利用保藏编号cctcc no:m 20807的植物乳杆菌zj316的发酵上清液,包括以下步骤:
[0011]
1)、将发酵上清先用大孔树脂xad-16吸附,然后依次用超纯水、30%甲醇、ph为7的50%甲醇进行洗脱,洗脱流速均为1
±
0.1ml/min,收集ph为7的50%甲醇对应的洗脱液;
[0012]
将所述洗脱液进行浓缩,得浓缩液;
[0013]
2)、将步骤1)所得的浓缩液经葡聚糖凝胶g25分离,获得g25-2组分;
[0014]
3)、将步骤2)所得的g25-2组分经反相高效液相色谱(rp-hplc)纯化:
[0015]
流动相a为体积浓度为0.05%的三氟乙酸水溶液;
[0016]
流动相b为体积浓度为0.05%的三氟乙酸乙腈溶液;
[0017]
洗脱剂由流动相a、流动相b组成,流速为3
±
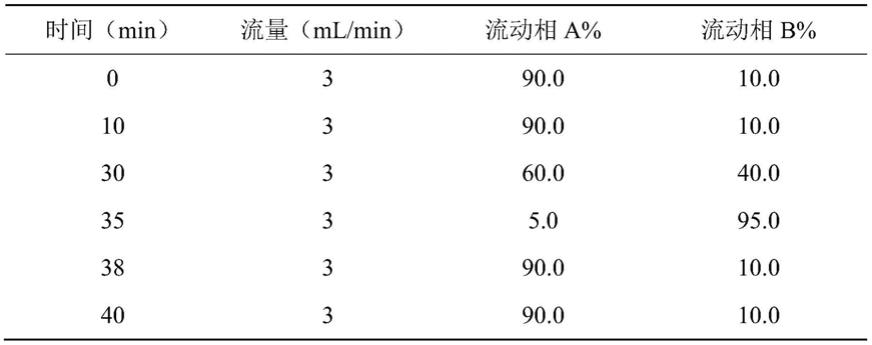
0.1ml/min,洗脱程序如下:
[0018]
rp-hplc洗脱程序
[0019][0020]
即:0~10min,流动相b 10%;10~30min,流动相b 10~40%;30~35min,流动相b 40~95%;35~38min,流动相b 95~10%;38~40min,流动相b 10%;
[0021]
收集出峰时间(起止时间)为23.9273~24.5716min的洗脱液,获得h4组分;将收集的洗脱液进行旋转蒸发仪浓缩,得吲哚-3-乳酸。
[0022]
作为本发明的从植物乳酸菌发酵上清中纯化吲哚-3-乳酸的方法的改进:所述步骤1)中,将洗脱液进行浓缩至原体积的9~11%,得浓缩液。
[0023]
作为本发明的从植物乳酸菌发酵上清中纯化吲哚-3-乳酸的方法的进一步改进:所述步骤1)为:
[0024]
5l植物乳杆菌zj316上清液经500g大孔树脂xad-16吸附(流速为1ml/min);然后依次用2l的超纯水、2l的30%甲醇、2l的ph为7的50%甲醇进行洗脱,洗脱流速均为1ml/min,收集ph为7的50%甲醇对应的洗脱液。
[0025]
作为本发明的从植物乳酸菌发酵上清中纯化吲哚-3-乳酸的方法的进一步改进,步骤2)包括:
[0026]
取步骤1)所得的浓缩液2ml,过0.22μm滤膜,上样至sephadex g25葡聚糖凝胶柱(柱高80cm,柱直径1.6cm);
[0027]
以超纯水为洗脱液,流速1
±
0.1ml/min,每隔3min接收1管洗脱液,收集t39、t40、t41、t42这4管洗脱液(为280nm波长下第2个色谱峰中吸光度值最高的4管);
[0028]
将洗脱液浓缩,得g25-2组分。
[0029]
作为本发明的从植物乳酸菌发酵上清中纯化吲哚-3-乳酸的方法的进一步改进,所述步骤2)的浓缩为:将洗脱液浓缩近干,用超纯水重溶至1ml,得g25-2组分。
[0030]
作为本发明的从植物乳酸菌发酵上清中纯化吲哚-3-乳酸的方法的进一步改进:所述步骤3)为:
[0031]
取步骤2)所得的g25-2组分(储存于4℃)500μl,用超纯水稀释至5ml后,上样至waters sunfire c18 prep(5μm 10
×
100mm),柱温为25℃,获得h4组分;
[0032]
所述步骤3)所得的h4组分浓缩近干,得吲哚-3-乳酸。
[0033]
作为本发明的从植物乳酸菌发酵上清中纯化吲哚-3-乳酸的方法的进一步改进:发酵上清的制备方法为:
[0034]
将保藏编号cctcc no:m 20807的植物乳杆菌zj316按3%(体积%)菌液接种量接种于mrs培养基中发酵(v/v),转速180rpm,37℃培养24h;发酵液离心(8000rpm,25min,4℃),获得上清(ph值约为3.79)。
[0035]
上述发酵可利用小型细菌型发酵罐发酵,即,将植物乳杆菌zj316按3%菌液接种于5lmrs中发酵。
[0036]
在本发明中,5l发酵上清液选用500g大孔树脂xad-16吸附,获得约2l 50%甲醇洗脱液;再经葡聚糖凝胶g25分离,获得g25-2组分;后经反相高效液相色谱(rp-hplc)制备纯化,rp-hplc型号及条件为waters sunfire c18(prep 5μm 10
×
100mm),流动相0.05%三氟乙酸/水(a)(v/v)和0.05%三氟乙酸/乙腈(b)(v/v)。
[0037]
在本发明中,植物乳杆菌zj316发酵上清液中吲哚-3-乳酸的含量是利用分析型hplc,色谱柱型号为waters sunfire c18(5μm 4.6
×
250mm)对一系列浓度梯度的吲哚-3-乳酸标品(sigma公司)进行分析,建立“峰面积—浓度”的线性关系,得回归方程。利用该方法测定植物乳杆菌zj316发酵上清液中吲哚-3-乳酸含量为43.14μg/ml,是目前已有报道的乳酸菌中吲哚-3-乳酸产量最高的植物乳杆菌之一。
[0038]
本发明所得的吲哚-3-乳酸具有抗菌特性,可有效抑制沙门氏菌、大肠杆菌、藤黄微球菌、金黄色葡萄球菌、铜绿假单胞菌等的生长。
[0039]
与现有技术相比,本发明具有如下技术优势:
[0040]
1、本发明利用hplc测定植物乳杆菌zj316发酵上清液中吲哚-3-乳酸的含量为43.14μg/ml,为目前报道的产量最高的植物乳杆菌之一。
[0041]
2、应用“大孔树脂xad-16—葡聚糖凝胶g25—反相高效液相色谱(rp-hplc)”三步法从植物乳杆菌zj316发酵上清液中获得吲哚-3-乳酸,纯度为99.00%,且具有广谱抗菌特性。
附图说明
[0042]
下面结合附图对本发明的具体实施方式作进一步详细说明。
[0043]
图1是吲哚-3-乳酸标准品回归方程图。
[0044]
图2是植物乳杆菌zj316发酵上清液中吲哚-3-乳酸的分离纯化及鉴定;
[0045]
图2中:a为葡聚糖凝胶g25分离;b为反相高效液相色谱(rp-hplc)分离纯化;c为lc-ms的液相色谱图;d为lc-ms的质谱图。
[0046]
图3是分析型高效液相色谱对吲哚-3-乳酸纯度的分析。
具体实施方式
[0047]
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:
[0048]
mrs液体培养基:无水葡萄糖20g,吐温-80 1ml,七水硫酸镁0.2g,硫酸锰0.05g,胰蛋白胨10g,酵母提取物5g,磷酸氢二钾2g,柠檬酸三铵2g,牛肉浸膏10g,无水乙酸钠5g,用超纯水溶解并定容至1l,用37.5%浓盐酸调节培养基ph=6。
[0049]
mrs固体培养基:在上述mrs液体培养基中加入2%(质量浓度)的细菌培养基琼脂,用37.5%浓盐酸调节培养基ph=6。
[0050]
实施例1:植物乳杆菌zj316发酵上清液中吲哚-3-乳酸含量测定
[0051]
(1)将植物乳杆菌zj316菌种划线于mrs固体培养基,37℃培养36h,挑取单菌落于10ml mrs液体培养基中37℃静置培养24h;
[0052]
(2)将步骤(1)所得培养液以3%接种量(体积浓度)接种于150ml mrs液体培养基中37℃继续静置培养24h;
[0053]
(3)将步骤(2)所得培养液以3%接种量(体积浓度)接种于5l mrs液体培养基中发酵。发酵条件:温度37℃,转速180rpm,时间24h。
[0054]
将所得的发酵液离心:8000rpm、25min、4℃,发酵上清液储存于4℃待用。
[0055]
(4)紫外检测条件:检测器型号waters 2498、检测器波长:280nm,柱温:25℃,进样量:30μl。对一系列浓度梯度(1.00、2.50、5.00、10.00、25.00、50.00、100.00μg/ml)的吲哚-3-乳酸标准品(购买于sigma公司)测定。建立出峰的峰面积与浓度的标准曲线,并计算得回归方程y=6687.8x+1905.4(r2=0.9994),如图1所示。
[0056]
(5)、分析型hplc测定步骤(3)所得的发酵上清中吲哚-3-乳酸含量,色谱柱型号为waters sunfire c18(5μm 4.6
×
250mm),取30μl步骤(3)所得的发酵上清液(储存于4℃)注入色谱柱内;洗脱剂由流动相a和流动相b组成:
[0057]
流动相a:0.05%三氟乙酸/水,即,为体积浓度为0.05%的三氟乙酸水溶液;
[0058]
流动相b:0.05%三氟乙酸/乙腈,即,为体积浓度为0.05%的三氟乙酸乙腈溶液;
[0059]
洗脱程序如下表1所述。
[0060]
表1、hplc洗脱程序
[0061][0062]
280nm紫外检测波长下,将与吲哚-3-乳酸标准品保留时间对应的色谱峰进行峰面积积分,所得峰面积为290395,即y=290395,代入图1回归方程y=6687.8x+1905.4(r2=
0.9994)计算,得植物乳杆菌zj316发酵上清液中吲哚-3-乳酸的含量为43.14μg/ml。
[0063]
实施例2:吲哚-3-乳酸的分离纯化及鉴定
[0064]
步骤一:大孔树脂xad-16吸附洗脱
[0065]
(1)5l植物乳杆菌zj316发酵上清液(实施例1步骤(3)所得)经500g大孔树脂xad-16吸附,流速为1ml/min;然后依次用2l的超纯水、30%甲醇、50%甲醇(37.5%浓盐酸调节ph=7),流速为1ml/min,收集ph=7的50%甲醇对应的洗脱液(约2l);
[0066]
(2)将步骤(1)所得的洗脱液(50%甲醇洗脱液)用旋转蒸发仪浓缩至200ml,得浓缩液;旋转蒸发条件:水浴温度37℃,压力30
±
5mbar,旋转速度60rpm。
[0067]
步骤二:葡聚糖凝胶柱sephadex g25分离
[0068]
(1)采用sephadex g25葡聚糖凝胶柱对步骤一所得的浓缩液进行柱分离(柱高80cm,柱直径1.6cm)。
[0069]
取步骤一所得的浓缩液2ml,过0.22μm滤膜,以超纯水为洗脱液,流速1ml/min,用自动接样器每隔3min接收1管洗脱液,即,每管g25洗脱液的体积为3ml;
[0070]
(2)用紫外分光光度计检测每管洗脱液在280nm波长下的吸光度值,记录并绘制“吸光度值—管数”,如图2a所示。收集管数“t39、t40、t41、t42”对应的样品,总体积为12ml,记为“g25-2”;
[0071]
(3)用旋转蒸发仪将步骤(2)所得的g25-2(12ml)浓缩近干,用超纯水重溶至1ml,得g25-2组分,储存于4℃待用;旋转蒸发条件:水浴温度37℃,压力30
±
5mbar,旋转速度60rpm。
[0072]
步骤三:反相高效液相色谱(rp-hplc)纯化
[0073]
取步骤二所得的g25-2组分(储存于4℃)500μl,用超纯水稀释至5ml。rp-hplc色谱柱型号为waters sunfire c18 prep(5μm 10
×
100mm),柱温为25℃,检测器型号waters2998,测器波长为280nm,流速为3ml/min。
[0074]
洗脱剂由流动相a和流动相b组成:
[0075]
流动相a:0.05%三氟乙酸/水,即,为体积浓度为0.05%的三氟乙酸水溶液;
[0076]
流动相b:0.05%三氟乙酸/乙腈,即,为体积浓度为0.05%的三氟乙酸乙腈溶液;
[0077]
洗脱程序如下表2所述。
[0078]
rp-hplc色谱图如图2b所示,收集其中出峰起止时间为23.9273~24.5716min出峰样品,所得记为“h4”。
[0079]
表2、rp-hplc洗脱程序
[0080]
[0081][0082]
步骤四:lc-ms分子量测定
[0083]
将步骤三所得的h4样品(约1.93ml),用旋转蒸发仪浓缩近干,用超纯水重溶至50μl,旋转蒸发条件:水浴温度37℃,压力30
±
5mbar,旋转速度60rpm,将浓缩所得物进行分子量鉴定。
[0084]
lc-ms仪器型号为安捷伦1200-6210,色谱柱型号为waters sunfire c18(5μm 4.6
×
250mm),柱温为30℃,离子源为esi,负离子模式,离子源气体温度为350℃,干燥气体流量为9l/min,雾化气压力为45psi,毛细管电压为3500v,碎裂电压为125v,质量数扫描范围为50-2000m/z,进样量为10μl。
[0085]
hplc洗脱剂由流动相a和流动相b组成:
[0086]
流动相a:0.1%甲酸/水,即,为体积浓度为0.1%的甲酸水溶液;
[0087]
流动相b:0.1%甲酸/乙腈,即,为体积浓度为0.1%的甲酸乙腈溶液;
[0088]
洗脱程序如下表3所述。
[0089]
流动相:0.1%甲酸水溶液(a)和乙腈(b),
[0090]
浓缩的h4样品的lc-ms图谱如图2c、2d所示,图2c为h4样品的液相色谱图,图2d为h4样品的质谱图,确定h4样品的分子量为205。
[0091]
因此,证明h4组分浓缩后所得物确实为吲哚-3-乳酸。
[0092]
表3、hplc洗脱程序
[0093][0094]
实施例3:分离纯化的吲哚-3-乳酸的纯度测定
[0095]
取实施例2步骤三的浓缩样品“h4”,按照实施例1步骤(5)中所述的分析型hplc检测条件和洗脱程序,得到吲哚-3-乳酸的色谱图,如图3所示。主成分峰(即,吲哚-3-乳酸)及2个杂峰的出峰起止时间(min)、峰面积(μv
·
s)、峰面积比例(%)如表4所示。根据峰面积归一化法,主成分峰(即,吲哚-3-乳酸)面积为a,主成分峰与其余杂峰峰面积总和为∑a,得吲哚-3-乳酸纯度(%)计算公式:
[0096]
吲哚-3-乳酸纯度(%)=a/∑a
×
100%。
[0097]
表4、hplc色谱峰的峰面积
[0098][0099]
因此,通过实施例2步骤一~三分离纯化得到:
[0100]
吲哚-3-乳酸纯度(%)=a/(a+a1+a2)
×
100%
[0101]
=691433/(691433+4028+2933)
×
100%
[0102]
=99.00%。
[0103]
实施例4:吲哚-3-乳酸的抗菌活性
[0104]
采用牛津杯法对实施例2步骤三纯化得到的吲哚-3-乳酸(即,浓缩h4,纯度99.00%)进行抗菌活性的测定。添加1%(体积浓度)的指示菌,150μl浓度10mg/ml吲哚-3-乳酸(1%乙腈水溶液溶解)。结果显示该植物乳杆菌zj316产生的吲哚-3-乳酸对指示菌的生长均有抑制作用(见表5),包括革兰氏阳性菌(藤黄微球菌、金黄色葡萄球菌、肉葡萄球菌、模仿葡萄球菌等),革兰氏阴性菌(大肠杆菌、甲/乙型副伤寒沙门氏菌、肠炎沙门氏菌、猪霍乱沙门氏菌、鼠伤寒沙门氏菌等)。
[0105]
表5、植物乳杆菌zj316产生的吲哚-3-乳酸的抑菌谱
[0106]
[0107][0108]
对比例1、将实施例1中的植物乳杆菌zj316,改成现有的其余植物乳杆菌菌株,具体如下表6所述,其余参照实施例1,检测植物乳杆菌发酵上清液中吲哚-3-乳酸的含量(μg/ml)。所得实验结果与本发明的对比如表6所述。
[0109]
表6
[0110][0111]
对比例2、将实施例1所得的植物乳杆菌zj316发酵上清液进行超滤离心,超滤离心的工艺参数具体如下:植物乳杆菌zj316上清液过0.22μm滤膜,用3kda超滤离心管(millipore amicon ultra)离心(4000g、4℃、30min),收集滤液。将超滤离心条件下的所得滤液,按照上述实施例3中所述方法进行纯度的检测,所得吲哚-3-乳酸的纯度约为40%。
[0112]
对比例3、将实施例2步骤一中的“大孔树脂xad-16”改成“大孔树脂xad-2”,其余等同于实施例2。所得吲哚-3-乳酸的纯度对应约为95%。
[0113]
对比例4-1、将实施例2步骤一中的“30%甲醇”改成“25%甲醇”,其余等同于实施例2。所得吲哚-3-乳酸的纯度约为83%。
[0114]
对比例4-2、将实施例2步骤一中的“30%甲醇”改成“35%甲醇”,其余等同于实施例2。所得吲哚-3-乳酸的纯度约为94%。
[0115]
对比例4-3、取消实施例2步骤一中的“30%甲醇”的使用,即,改成“用2l的超纯水、50%甲醇(ph=7)”,其余等同于实施例2。所得吲哚-3-乳酸的纯度约为35%。
[0116]
对比例4-4、将实施例2步骤一中的“50%甲醇(ph=7)”改成“50%甲醇”,即,取消对50%甲醇的ph调节,其余等同于实施例2。所得吲哚-3-乳酸的纯度约为75%。
[0117]
对比例5、将实施例2步骤二中的“葡聚糖凝胶柱sephadex g25”改成“葡聚糖凝胶柱sephadex g50柱”或“葡聚糖凝胶柱sephadex g15柱”,其余等同于实施例2。改用“葡聚糖凝胶柱sephadex g50柱”,无法得到与图2a类似的色谱图,只含有1个样品峰,即无法得到组分g25-2;改用“葡聚糖凝胶柱sephadex g15柱”,可得到与图2a类似的色谱图,含有2个样品
峰,即,可得到组分g25-2,其余等同于实施例2。所得吲哚-3-乳酸的纯度对应约为78%。
[0118]
对比例6、将实施例2步骤二中的葡聚糖凝胶柱sephadex g25对应的流速“1ml/min”改成“0.5ml/min”或“1.5ml/min”,其余等同于实施例2。所得吲哚-3-乳酸的纯度对应约为98%、70%。
[0119]
对比例7、将实施例2步骤三中的反相高效液相色谱(rp-hplc)所用的色谱柱“waters sunfire c18 prep(5μm 10
×
100mm)”改成“ymc-pack pro c18(5μm 20
×
150mm)”,其余等同于实施例2。则无法得到与图2b类似的色谱图,即,无法得到单峰组分h4。
[0120]
对比例8、将实施例2步骤三中的反相高效液相色谱(rp-hplc)洗脱程序改为如下:
[0121]
0~10min,流动相b 5%;10~30min,流动相b 5~50%;30~35min,流动相b 50~95%;35~38min,流动相b 95~5%;38~40min,流动相b 5%;流量为3ml/min。
[0122]
或0~10min,流动相b 10%;10~30min,流动相b 10~50%;30~35min,流动相b 50~95%;35~38min,流动相b 95~10%;38~40min,流动相b 10%;流量为3ml/min。
[0123]
或0~10min,流动相b 5%;10~30min,流动相b 5~40%;30~35min,流动相b 40~95%;35~38min,流动相b 95~5%;38~40min,流动相b 5%;流量为3ml/min。
[0124]
或0~10min,流动相b 10%;10~30min,流动相b 10~40%;30~35min,流动相b 40~95%;35~38min,流动相b 95~10%;38~40min,流动相b 10%;流量为2ml/min。
[0125]
或0~10min,流动相b 10%;10~30min,流动相b 10~40%;30~35min,流动相b 40~95%;35~38min,流动相b 95~10%;38~40min,流动相b 10%;流量为4ml/min。
[0126]
其余等同于实施例2。所得吲哚-3-乳酸的纯度对应约为92%、96%、96%、90%、82%。
[0127]
最后,还需要注意的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。