1.本发明涉及液晶化合物合成技术领域,尤其涉及一种含多氟萘的液晶化合物及其制备方法。
背景技术:
2.近几十年来,液晶材料在显示器方面的应用得到了飞速发展,随着集成电路和液晶设备的改进提高,人们对液晶材料的性能要求也越来越高。液晶材料在现代生活中扮演着不可或缺的角色,手表、计算机、仪器仪表,以及手机和液晶电视、电脑等都离不开液晶显示的支持。
3.液晶材料通常具有低粘度、良好的混溶性、电阻率高、以及化学稳定性、热稳定性和光稳定性好的优点,同时还具有较宽的向列相温度和光学、电化学各向异性等特点。通常,需要将多种不同单一液晶材料按照一定的比例混合以达到液晶显示的要求,因此,对于某些性能优良的液晶新材料的研究成为新的突破。
4.在已知的液晶化合物中,多氟类液晶化合物具有较大的负介电各向异性,粘度低、电阻率高以及稳定性好等优点,含多氟类化合物混配成的液晶材料能够满足化学稳定性高、粘度低以及电荷保持率高等要求,在各种显示模式的液晶元件中具有很大用途。因此,研发新的液晶化合物,使显示器件的性能更加优异,对于推动液晶化合物及显示器件的性能不断向前发展具有重要意义。
技术实现要素:
5.鉴于此,本发明提供了一种含多氟萘的液晶化合物及其制备方法。
6.为解决上述技术问题,本发明提供的技术方案是:
7.一方面,本发明提供了一种含多氟萘的液晶化合物,其结构如式(i)所示:
[0008][0009]
其中,r1、r3为c1‑
c9的直链烷基或环戊基;r2为
‑
h或
‑
f。
[0010]
相对于现有技术,本发明提供了一种新型结构的含多氟萘的液晶化合物,多氟萘的引入可显著提高液晶化合物的分子极性,使其具有较宽的向列相温度范围,较大的介电各向异性δε,更合适的光学各项异性δn等优点,同时,还具有良好的热稳定性,较高的清亮点cp,且与其他液晶化合物相溶性优异等优点,作为显示用液晶材料能够改善液晶组合物的响应速度和驱动电压,可以用作垂直配向方式、ips等液晶显示元件的构成部件,具有较高的推广应用价值。
[0011]
优选的,r1,r3为c1‑
c5的直链烷基或环戊基。
[0012]
优选的液晶化合物具有较低的旋转粘度,较大的δε和更合适的δn,还具有较高的清亮点,以及优良的液晶互溶性,有利于改善液晶组合物的工作温度;此外,还具有良好的热稳定性、化学稳定性、光学稳定性,从而有效降低驱动电压,提高液晶显示器件的响应速度。
[0013]
另一方面,本发明还提供了一种上述含多氟萘的液晶化合物的制备方法,包括如下步骤:
[0014]
步骤a,将式(v)所述的烷基环己基甲酸进行还原,得式(iv)所示的烷基环己基甲醇;然后将所述烷基环己基甲醇与2,3
‑
二氟苯酚进行光延反应,得式(iii)所示的烷基环己基甲氧基氟苯;
[0015][0016]
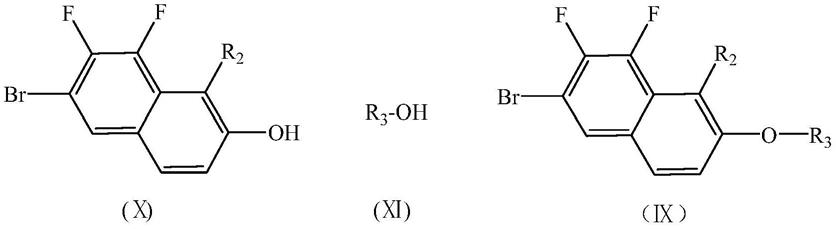
步骤b,将式(
ⅹ
)所示的含氟溴萘酚与式(
ⅺ
)所示烷基醇进行光延反应,得式(
ⅸ
)所示的含氟溴萘;
[0017][0018]
步骤c,将式(
ⅸ
)所示的含氟溴萘与锂试剂进行亲核取代反应,得式(
ⅷ
)所示的萘锂试剂,然后将所述萘锂试剂与1,4
‑
环己二酮单乙二醇缩酮进行亲核加成反应,再经水解、脱水、加氢和脱乙二醇反应,得式(
ⅵ
)所示的萘环己基酮;
[0019][0020]
步骤d,将式(iii)所示的烷基环己基甲氧基氟苯与锂试剂进行亲核取代反应后,再与式(
ⅵ
)所示的萘环己基酮进行亲核加成反应,然后经水解、脱水和加氢反应,得式(i)所示的含多氟萘的液晶化合物。反应方程式如下:
[0021][0022]
目前已经有较多含环己基的氟苯类液晶化合物的合成方法的报道,传统的合成含环己基的氟苯类液晶化合物的方法一般是采用烷基环己基醇类化合物与磺酰氯化合物进行磺酰化反应后,再与含氟苯酚类化合物进行反应制备得到目标产物。这一工艺路线需要用到对环境和人体危害较大的磺酰氯类化合物,操作安全隐患较大,且产品收率较低。
[0023]
本发明利用烷基环己基甲醇与2,3
‑
二氟苯酚,以及含氟溴萘酚与烷基醇通过光延反应分别得到式(iii)所示的烷基环己基甲氧基氟苯和式(
ⅸ
)所示的含氟溴萘,然后含氟溴萘与1,4
‑
环己二酮单乙二醇缩酮反应制备式(
ⅵ
)所示的萘环己基酮,之后将烷基环己基甲氧基氟苯与萘环己基酮通过亲核加成等反应,合成了既含环己基又含有多氟萘的液晶化合物。该合成路线操作安全,采用光延反应避免了使用磺酰氯类化合物,以及卤代物偶联,反应中杂质产生少,利于获得高纯度液晶单体,对于制备品质要求较高的液晶显示材料具有重要价值。
[0024]
优选的,步骤a具体步骤包括:
[0025]
步骤1,将式(v)所述的烷基环己基甲酸溶于第一有机溶剂中,于0℃~30℃加入还原剂,反应1h~4h,得式(iv)所示的烷基环己基甲醇;
[0026]
步骤2,将式(iv)所示的烷基环己基甲醇、2,3
‑
二氟苯酚和三苯基膦溶于甲苯中,在
‑
10℃~0℃加入偶氮二甲酸酯的甲苯溶液,升温至20℃~30℃,反应1h~3h,得式(iii)所示的烷基环己基甲氧基氟苯。
[0027]
式(v)所示的烷基环己基甲酸原料易得,成本较低,由其通过简单的还原反应制备得到式(iv)所示的烷基环己基甲醇,可以有效降低生产成本。且后续通过光延反应制备得到烷基环己基甲氧基氟苯,避免了使用对环境和人体危害较大的磺酰氯类化合物,所使用的溶剂也较为环保,操作易控,反应条件较为温和,危险因素较少,且烷基环己基甲氧基氟苯的收率较高,有较高的推广应用价值。
[0028]
更优选的,上述步骤1中,所述还原剂为氢化铝锂、氢化二异丁基铝或双(2
‑
甲氧基乙氧基)氢化铝钠中至少一种。
[0029]
更优选的,上述步骤1中,所述第一有机溶剂为四氢呋喃、2
‑
甲基四氢呋喃、甲基叔丁基醚、环己烷或甲苯中至少一种。
[0030]
更优选的,上述步骤1中,所述式(v)所述的烷基环己基甲酸与还原剂的摩尔比为1:1~1.2。
[0031]
更优选的,上述步骤1中,还原反应1h~4h后,用氢氧化钠溶液淬灭反应,用甲苯萃取,合并有机相,用水洗涤,无水硫酸钠干燥,减压蒸馏脱除溶剂,得烷基环己基甲醇粗品,
然后用乙酸乙酯和正庚烷的混合溶液溶解烷基环己基甲醇粗品,过硅胶柱,浓缩,得式(iv)所示的烷基环己基甲醇产品。
[0032]
进一步优选的,上述步骤1中,所述乙酸乙酯和正庚烷的体积比为1:4。
[0033]
采用优选的反应条件,如反应温度、还原剂、溶剂以及还原剂的加入量等,有利于提高原料的利用率,降低副反应的发生,从而提高烷基环己基甲醇的收率和纯度,进而有利于提高下游产品的收率和纯度。
[0034]
更优选的,上述步骤2中,所述式(iv)所示的烷基环己基甲醇、2,3
‑
二氟苯酚、三苯基膦与偶氮二甲酸酯的摩尔比为1:1:1.1~1.3:1.1~1.3。
[0035]
更优选的,上述步骤2中,所述偶氮二甲酸酯为偶氮二甲酸二甲酯、偶氮二甲酸二乙酯或偶氮二甲酸二异丙酯中至少一种。
[0036]
更优选的,上述步骤2中,所述偶氮二甲酸酯的甲苯溶液中偶氮二甲酸酯与甲苯的摩尔体积比为1:1~1.1,其中,体积的单位是升。
[0037]
更优选的,上述步骤2中,升温反应1h~3h后,加入水淬灭反应,经分液,萃取,水洗,无水硫酸钠干燥,浓缩,用乙酸乙酯和正庚烷混合溶液溶解粗品,过硅胶柱后浓缩,正庚烷结晶,得式(iii)所示的烷基环己基甲氧基氟苯。
[0038]
进一步优选的,上述步骤2中,所述乙酸乙酯和正庚烷的体积比为1:4。
[0039]
优选的光延反应条件,有利于提高原料的转化率,降低副反应的发生,从而提高烷基环己基甲氧基氟苯产品的收率和纯度。
[0040]
优选的,步骤b具体步骤包括:
[0041]
将所述式(
ⅹ
)所示含氟溴萘酚、式(
ⅺ
)所示烷基醇和三苯基膦溶于甲苯中,在
‑
10℃~0℃加入偶氮二甲酸酯的甲苯溶液,升温至20℃~30℃,反应1h~3h,得式(
ⅸ
)所示的含氟溴萘。
[0042]
优选的,步骤b中,所述式(
ⅹ
)所示的含氟溴萘酚、式(
ⅺ
)所示的烷基醇、三苯基膦与偶氮二甲酸酯的摩尔比为1:1:1.1~1.3:1.1~1.3。
[0043]
优选的,步骤b中,所述偶氮二甲酸酯为偶氮二甲酸二甲酯、偶氮二甲酸二乙酯或偶氮二甲酸二异丙酯中至少一种。
[0044]
优选的,步骤b中,所述偶氮二甲酸酯的甲苯溶液中偶氮二甲酸酯与甲苯的摩尔体积比为1:1~1.1,其中,体积的单位是升。
[0045]
优选的,上述步骤b中,升温反应1h~3h后,加入水淬灭反应,经分液,萃取,水洗,无水硫酸钠干燥,浓缩,用乙酸乙酯和正庚烷混合溶液溶解粗品,过硅胶柱后浓缩,正庚烷结晶,得式(
ⅸ
)所示的含氟溴萘。
[0046]
进一步优选的,上述步骤b中,所述乙酸乙酯和正庚烷的体积比为1:4。
[0047]
优选的,步骤c中,所述亲核取代反应的反应温度为
‑
100℃~
‑
60℃,反应时间为0.5h~4h。
[0048]
优选的,步骤c中,所述亲核加成反应的反应温度为
‑
100℃~
‑
60℃,反应时间为0.5h~4h。
[0049]
优选的,步骤c中,所述锂试剂为甲基锂、正丁基锂、仲丁基锂、叔丁基锂或正己基锂中至少一种。
[0050]
优选的,步骤c中,所述1,4
‑
环己二酮单乙二醇缩酮、式(
ⅸ
)所示的含氟溴萘和锂
试剂的摩尔比为1:1~1.2:1~1.2。
[0051]
优选的,步骤d中,所述亲核取代反应的温度为
‑
100℃~
‑
60℃,反应时间为0.5h~4h。
[0052]
优选的,步骤d中,所述亲核加成反应的反应温度为
‑
100℃~
‑
60℃,反应时间为0.5h~4h。
[0053]
优选的,步骤d中,所述锂试剂为甲基锂、正丁基锂、仲丁基锂、叔丁基锂或正己基锂中至少一种。
[0054]
优选的,步骤d中,所述式(
ⅵ
)所示的萘环己基酮、式(iii)所示的烷基环己基甲氧基氟苯与锂试剂的摩尔比为1:1~1.2:1~1.2。
[0055]
优选的,步骤c和步骤d中,脱水反应的温度均为100℃~110℃,时间均为0.5h~4h。
[0056]
优选的,步骤c,水解反应采用质量浓度为8wt%~12wt%的氯化铵水溶液进行水解。
[0057]
优选的,步骤d中,水解反应采用质量浓度为8wt%~12wt%的乙酸水溶液进行水解。
[0058]
优选的,步骤c和步骤d中,均采用酸性脱水剂进行脱水。
[0059]
将亲核加成反应的产物在酸性条件进行水解后,静置分层,乙酸乙酯萃取,合并有机相,水洗至中性,干燥,浓缩脱除溶剂后,将产物溶于甲苯溶剂中,加入酸性脱水剂,于100℃~110℃脱水0.5h~4h,分液,收集有机相,水洗至中性,浓缩脱除溶剂,得脱水产物。
[0060]
进一步优选的,所述酸性脱水剂为对甲基苯磺酸。
[0061]
优选的,步骤c和步骤d中,加氢反应的温度均为10℃~50℃,压力均为0.5mpa~2.0mpa,时间均为2h~4h。
[0062]
优选的,所述加氢反应具体为:将上述脱水产物加入第二有机溶剂中,以pd/c或雷尼镍作为加氢催化剂,在10℃~50℃,压力0.5mpa~2.0mpa条件下,反应2h~4h,过滤,浓缩,柱层析,得加氢产物。
[0063]
更优选的,所述第二有机溶剂为四氢呋喃、异丙醇、甲醇或乙醇中至少一种。
[0064]
更优选的,上述柱层析采用体积比为1:1的乙酸乙酯和正庚烷的混合溶液。
[0065]
优选的加氢反应条件,可提高原料的转化率,提高加氢反应产物的收率。
[0066]
优选的,步骤c中,脱乙二醇保护的反应温度为0℃~50℃,反应时间为0.5h~4h。
[0067]
更优选的,脱乙二醇保护反应具体为:将上述加氢产物溶于第三有机溶剂中,加入酸溶液,于0℃~50℃,反应0.5h~4h,分液,萃取,水洗干燥,浓缩,正庚烷重结晶,得式(
ⅵ
)所示的萘环己基酮。
[0068]
进一步优选的,脱乙二醇保护反应中,所述加氢产物与第三有机溶剂的摩尔体积比为1:1.8~2,其中,体积的单位是升。
[0069]
进一步优选的,脱乙二醇保护反应中,所述第三有机溶剂为四氢呋喃、甲苯、甲醇或乙醇中至少一种。
[0070]
进一步优选的,脱乙二醇保护反应中,所述酸溶液为甲酸、三氟乙酸、乙酸、磷酸或硫酸中至少一种。
[0071]
进一步优选的,脱乙二醇保护反应中,所述酸溶液的质量浓度为8wt%~12wt%。
[0072]
进一步优选的,脱乙二醇保护反应中,所述加氢产物与上述酸溶液的摩尔体积比为1:1~1.2,其中,体积的单位是升。
[0073]
优选的脱水、加氢和脱保护等反应条件,有利于提高各步反应的产物的收率和纯度。
[0074]
本发明提供了一种新颖、操作简单且安全性较高的含多氟萘的液晶化合物的制备方法,产品收率可达86%以上,纯度可达99.5%以上,且整个制备过程中未使用危险性较高的磺酰氯类化合物和石油醚溶剂,适用于工业化生产应用,推广价值极高。
具体实施方式
[0075]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0076]
为了更好的说明本发明,下面通过实施例作进一步的举例说明。
[0077]
实施例1
[0078]
本实施例提供一种含多氟萘的液晶化合物的制备方法,包括如下步骤:
[0079]
(1)反式
‑4‑
丙基环己基甲醇(ⅳ)的制备:
[0080]
将反式
‑4‑
丙基环己基甲酸17.0g(0.1mol)和102ml甲苯加入500ml三口瓶中,冰浴控温,于0~5℃滴加28.9g双(2
‑
甲氧基乙氧基)氢化铝钠的甲苯溶液(双(2
‑
甲氧基乙氧基)氢化铝钠的质量百分含量为70%,0.1mol),然后升温至25℃反应3h,于25℃条件下滴入66.7ml 20wt%的氢氧化钠水溶液结束反应。然后静置分层,分取有机层,用甲苯萃取水层,将有机相合并,用水洗涤,无水硫酸钠干燥,减压蒸馏脱除溶剂,用31ml乙酸乙酯和124ml正庚烷溶解粗品,过15.6g硅胶柱,收集,浓缩,得反式
‑4‑
丙基环己基甲醇14g,白色固体,收率90%,gc纯度99.8%。
[0081]
(2)1
‑
((反式
‑4‑
丙基环己基)甲氧基)
‑
2,3
‑
二氟苯(ⅲ)的制备:
[0082]
在500ml三口瓶中安装好温度计、机械搅拌和恒压滴液漏斗,加入甲苯150ml、反式
‑4‑
丙基环己基甲醇15.6g(0.1mol)、2,3
‑
二氟苯酚13g(0.1mol)、三苯基膦28.9g(0.11mol),搅拌混合均匀,氮气保护下,冷却至
‑
10℃,滴加偶氮二甲酸二甲酯16.1g(0.11mol)与111.2ml甲苯的混合溶液,滴加过程温度控制为
‑
10~0℃,滴加结束后升温至25℃,保温2h,保温结束后于25℃加入24.5g水,保温搅拌3h,分液,向水相中加入甲苯萃取,合并有机相,水洗,无水硫酸钠干燥,浓缩,用54ml乙酸乙酯和216ml正庚烷溶解粗品,过27g硅胶柱,收集,浓缩,加入正庚烷搅拌结晶0.5h,得1
‑
((反式
‑4‑
丙基环己基)甲氧基)
‑
2,3
‑
二氟苯白色固体24.2g,收率90%,gc纯度99.5%。
[0083]
(3)3
‑
溴
‑
1,2,8
‑
三氟
‑7‑
丙氧基萘(
ⅸ
)的制备:
[0084]
在500ml三口瓶中安装好温度计、机械搅拌和恒压滴液漏斗,加入甲苯150ml、6
‑
溴
‑
1,7,8
‑
三氟
‑2‑
萘酚27.7g(0.1mol)、正丙醇6.0g(0.1mol)、三苯基膦28.9g(0.11mol),搅拌混合均匀,氮气保护下,冷却至
‑
10℃,滴加偶氮二甲酸二甲酯16.1g(0.11mol)与111.2ml甲苯的混合溶液,滴加过程温度控制为
‑
10~0℃,滴加结束后升温至25℃,保温2h,保温结束后于25℃加入24.5g水,保温搅拌3h,分液,向水相中加入甲苯萃取,合并有机相,水洗,无水硫酸钠干燥,浓缩,用64ml乙酸乙酯和256ml正庚烷溶解粗品,过32g硅胶柱,收
集,浓缩,加入正庚烷搅拌结晶0.5h,得3
‑
溴
‑
1,2,8
‑
三氟
‑7‑
丙氧基萘白色固体28.7g,收率90%,gc纯度99.5%。
[0085]
(4)8
‑
(3,4,5
‑
三氟
‑6‑
丙氧基萘
‑2‑
基)
‑
1,4
‑
二氧杂螺[4.5]癸烷(
ⅶ
)的制备:
[0086]
在1000ml三口瓶中安装好温度计、机械搅拌装置并通入氮气,向体系中加入3
‑
溴
‑
1,2,8
‑
三氟
‑7‑
丙氧基萘31.9g(0.1mol)和159.5ml四氢呋喃,搅拌混合均匀,将体系降温至
‑
90℃,然后向体系中保温滴加浓度为2.7mol/l正丁基锂的己烷溶液37ml(0.1mol),滴加过程中控温
‑
90℃,滴加结束后保温3h,于
‑
90℃向体系中滴加1,4
‑
环己二酮单乙二醇缩酮15.6g(0.1mol)和50ml四氢呋喃的混合溶液,滴毕,保温反应3h,自然升温至0℃搅拌,之后将体系中倒入53ml 10wt%的氯化铵水溶液中进行水解,静置分层,乙酸乙酯萃取,合并有机相,水洗至中性,干燥,浓缩脱除溶剂,将所得固体溶于239ml甲苯中,加入1g对甲基苯磺酸,安装脱水装置,升温至110℃后,回流分水反应4h,降至室温,倒入分液漏斗中分液,加水水洗至中性,干燥,浓缩脱干溶剂,向其中加入235ml乙醇,加入1g雷尼镍,于温度20℃、压力2mpa条件下进行催化氢化,反应4h后取样分析,加氢反应完毕,过滤,浓缩,用114ml乙酸乙酯和114ml正庚烷溶解粗品,过硅胶柱进行柱层析,收集,浓缩,得8
‑
(3,4,5
‑
三氟
‑6‑
丙氧基萘
‑2‑
基)
‑
1,4
‑
二氧杂螺[4.5]癸烷33.4g,收率88%,gc纯度99.4%。
[0087]
(5)4
‑
(3,4,5
‑
三氟
‑6‑
丙氧基萘
‑2‑
基)环己酮(
ⅵ
)的制备:
[0088]
取1000ml三口瓶中安装好温度计、机械搅拌装置,加入8
‑
(3,4,5
‑
三氟
‑6‑
丙氧基萘
‑2‑
基)
‑
1,4
‑
二氧杂螺[4.5]癸烷38g(0.1mol)和190ml甲苯,搅拌混合均匀,加入10wt%硫酸水溶液100ml,于50℃搅拌反应0.5h,分液,甲苯萃取水相,合并有机相,水洗,浓缩至干,加入正庚烷重结晶,得4
‑
(3,4,5
‑
三氟
‑6‑
丙氧基萘
‑2‑
基)环己酮30.6g,收率91%,纯度99.3%。
[0089]
(6)3
‑
(反式
‑4‑
(2,3
‑
二氟
‑4‑
((反式
‑4‑
丙基环己基)甲氧基)苯基)环己基)
‑
1,2,8
‑
三氟
‑7‑
丙氧基萘(ⅰ)的制备:
[0090]
在1000ml三口瓶中安装好温度计、机械搅拌装置并通入氮气,向体系中加入1
‑
((反式
‑4‑
丙基环己基)甲氧基)
‑
2,3
‑
二氟苯26.8g(0.1mol)和134ml四氢呋喃,搅拌混和均匀,将体系降温至
‑
90℃,然后向体系中保温滴加浓度为2.7mol/l正丁基锂的己烷溶液37ml(0.1mol),滴加过程中控温
‑
90℃,滴加结束保温反应4h,于
‑
90℃向体系中滴加4
‑
(3,4,5
‑
三氟
‑6‑
丙氧基萘
‑2‑
基)环己酮33.6g(0.1mol)和67ml四氢呋喃的混合溶液,滴毕,保温反应4h,自然升温至0℃搅拌,之后将体系倒入100ml 10wt%的乙酸水溶液中水解,静置分层,乙酸乙酯萃取,合并有机相,水洗有机相至中性,干燥,浓缩脱干溶剂后,将所得固体溶于242ml甲苯中,向其中加入1g对甲基苯磺酸,安装脱水装置,升温至110℃后回流分水4h,降温至室温,倒入分液漏斗中,加水水洗至中性,干燥,浓缩脱干溶剂后,向其中加入352ml乙醇,加入5%pd/c催化剂1g,于温度30℃、压力2mpa条件下进行催化氢化,反应4h后取样分析,加氢完毕,经过滤、浓缩,用176ml乙酸乙酯和176ml正庚烷溶解粗品,过硅胶柱进行柱层析,收集,浓缩,得3
‑
(反式
‑4‑
(2,3
‑
二氟
‑4‑
((反式
‑4‑
丙基环己基)甲氧基)苯基)环己基)
‑
1,2,8
‑
三氟
‑7‑
丙氧基萘52.3g,收率89%,gc纯度99.6%。
[0091][0092]1h nmr(400mhz,cd3cl),δ7.43(d,1h),7.16
‑
7.02(m,2h),6.74(d,1h),6.15(d,1h),4.28(t,2h),3.90(d,2h),2.88
‑
2.57(d,2h),2.37
–
2.22(m,6h),2.10
–
1.52(m,8h),1.46
–
1.33(t,3h),1.27
–
1.12(m,10h),0.87(t,3h)。
[0093]
实施例2
[0094]
本实施例提供一种含多氟萘的液晶化合物的制备方法,包括如下步骤:
[0095]
(1)反式
‑4‑
环戊基环己基甲醇(ⅳ)的制备:
[0096]
将反式
‑4‑
环戊基环己基甲酸19.6g(0.1mol)和100ml四氢呋喃加入500ml三口瓶中,冰浴控温,于0~5℃滴加氢化铝锂的四氢呋喃溶液110ml(110ml四氢呋喃中含0.11mol氢化铝锂),保温搅拌1h,于0~5℃条件下滴入66.7ml 20wt%的氢氧化钠水溶液结束反应。然后静置分层,分取有机层,用甲苯萃取水层,将有机层合并,用水洗涤,无水硫酸钠干燥,减压蒸馏脱除溶剂,用37ml乙酸乙酯和148ml正庚烷溶解粗品,过18.3g硅胶柱,收集,浓缩,得反式
‑4‑
环戊基环己基甲醇16.8g,收率92%,gc纯度99.5%。
[0097]
(2)1
‑
((反式
‑4‑
环戊基环己基)甲氧基)
‑
2,3
‑
二氟苯(ⅲ)的制备:
[0098]
在500ml三口瓶中安装好温度计、机械搅拌和恒压滴液漏斗,加入甲苯150ml、反式
‑4‑
环戊基环己基甲醇18.2g(0.1mol)、2,3
‑
二氟苯酚13g(0.1mol)、三苯基膦31.5g(0.12mol),搅拌混合均匀,氮气保护下,冷却至
‑
10℃,滴加偶氮二甲酸二乙酯20.9g(0.12mol)与121.5ml甲苯的混合溶液,滴加过程温度控制为
‑
10~0℃,滴加结束后升温至25℃,保温2h,保温结束后于25℃加入24.5g水,保温搅拌2h,分液,向水相中加入甲苯萃取,合并有机相,水洗,无水硫酸钠干燥,浓缩,用59ml乙酸乙酯和236ml正庚烷溶解粗品,过29.5g硅胶柱,收集,浓缩,加入正庚烷搅拌结晶0.5h,得1
‑
((反式
‑4‑
环戊基环己基)甲氧基)
‑
2,3
‑
二氟苯26.8g,收率91%,gc纯度99.4%。
[0099]
(3)3
‑
溴
‑
1,2,8
‑
三氟
‑7‑
乙氧基萘(
ⅸ
)的制备:
[0100]
在500ml三口瓶中安装好温度计、机械搅拌和恒压滴液漏斗,加入甲苯150ml、6
‑
溴
‑
1,7,8
‑
三氟
‑2‑
萘酚27.7g(0.1mol)、乙醇4.6g(0.1mol)、三苯基膦31.5g(0.12mol),搅拌混合均匀,氮气保护下,冷却至
‑
10℃,滴加偶氮二甲酸二乙酯20.9g(0.12mol)与121.4ml甲苯的混合溶液,滴加过程温度控制为
‑
10~0℃,滴加结束后升温至25℃,保温2h,保温结束后于25℃加入24.5g水,保温搅拌3h,分液,向水相中加入甲苯萃取,合并有机相,水洗,无水硫酸钠干燥,浓缩,用61ml乙酸乙酯和244ml正庚烷溶解粗品,过30.5g硅胶柱,收集,浓缩,加入正庚烷搅拌结晶0.5h,得3
‑
溴
‑
1,2,8
‑
三氟
‑7‑
乙氧基萘白色固体27.5g,收率90%,gc纯度99.5%。
[0101]
(4)8
‑
(3,4,5
‑
三氟
‑6‑
乙氧基萘
‑2‑
基)
‑
1,4
‑
二氧杂螺[4.5]癸烷(
ⅶ
)的制备:
[0102]
在1000ml三口瓶中安装好温度计、机械搅拌装置并通入氮气,向体系中加入3
‑
溴
‑
1,2,8
‑
三氟
‑7‑
乙氧基萘36.6g(0.12mol)和183ml四氢呋喃,搅拌混合均匀,将体系降温至
‑
75℃,然后向体系中保温滴加浓度为2.5mol/l正己基锂的己烷溶液48ml(0.12mol),滴加过程中控温
‑
75℃,滴加结束后保温1h,于
‑
75℃向体系中滴加1,4
‑
环己二酮单乙二醇缩酮15.6g(0.1mol)和50ml四氢呋喃的混合溶液,滴毕,保温反应2h,自然升温至0℃搅拌,之后将体系中倒入53ml 10wt%的氯化铵水溶液中进行水解,静置分层,乙酸乙酯萃取,合并有机相,水洗至中性,干燥,浓缩脱除溶剂,将所得固体溶于275ml甲苯中,加入1g对甲基苯磺酸,安装脱水装置,升温至110℃后,回流分水反应2h,降至室温,倒入分液漏斗中分液,加水水洗至中性,干燥,浓缩脱干溶剂,向其中加入262ml乙醇,加入1g 5%pd/c催化剂,于温度30℃、压力1mpa条件下进行催化氢化,反应2h后取样分析,加氢反应完毕,过滤,浓缩,用109ml乙酸乙酯和109ml正庚烷溶解粗品,过硅胶柱进行柱层析,收集,浓缩,得8
‑
(3,4,5
‑
三氟
‑6‑
乙氧基萘
‑2‑
基)
‑
1,4
‑
二氧杂螺[4.5]癸烷31.5g,收率86%,gc纯度99.5%。
[0103]
(5)4
‑
(3,4,5
‑
三氟
‑6‑
乙氧基萘
‑2‑
基)环己酮(
ⅵ
)的制备:
[0104]
取1000ml三口瓶中安装好温度计、机械搅拌装置,加入8
‑
(3,4,5
‑
三氟
‑6‑
乙氧基萘
‑2‑
基)
‑
1,4
‑
二氧杂螺[4.5]癸烷36.6g(0.1mol)和180ml甲苯,搅拌混合均匀,加入10wt%乙酸水溶液100ml,于30℃搅拌反应2h,分液,甲苯萃取水相,合并有机相,水洗,干燥,浓缩至干,加入正庚烷重结晶,得4
‑
(3,4,5
‑
三氟
‑6‑
乙氧基萘
‑2‑
基)环己酮29g,收率90%,纯度99.3%。
[0105]
(6)3
‑
(反式
‑4‑
(2,3
‑
二氟
‑4‑
((反式
‑4‑
环戊基环己基)甲氧基)苯基)环己基)
‑
1,2,8
‑
三氟
‑7‑
乙氧基萘(ⅰ)的制备:
[0106]
在1000ml三口瓶中安装好温度计、机械搅拌装置并通入氮气,向体系中加入1
‑
((反式
‑4‑
环戊基环己基)甲氧基)
‑
2,3
‑
二氟苯35.3g(0.12mol)和134ml四氢呋喃,搅拌混和均匀,将体系降温至
‑
70℃,然后向体系中保温滴加浓度为2.5mol/l正己基锂的己烷溶液48ml(0.12mol),滴加过程中控温
‑
70℃,滴加结束保温反应1h,于
‑
70℃向体系中滴加4
‑
(3,4,5
‑
三氟
‑6‑
乙氧基萘
‑2‑
基)环己酮32.2g(0.1mol)和64ml四氢呋喃的混合溶液,滴毕,保温反应1h,自然升温至0℃搅拌,之后将体系倒入100ml 10wt%的乙酸水溶液中水解,静置分层,乙酸乙酯萃取,合并有机相,水洗有机相至中性,浓缩脱干溶剂后,将所得固体溶于370ml甲苯中,向其中加入1g对甲基苯磺酸,安装脱水装置,升温至110℃后回流分水3h,降温至室温,倒入分液漏斗中,加水水洗至中性,干燥,浓缩脱干溶剂后,向其中加入359ml甲醇,加入雷尼镍催化剂1g,于温度30℃、压力1mpa条件下进行催化氢化,反应2h后取样分析,加氢完毕,经过滤、浓缩,用180ml乙酸乙酯和180ml正庚烷溶解粗品,过硅胶柱进行柱层析,收集,浓缩,得3
‑
(反式
‑4‑
(2,3
‑
二氟
‑4‑
((反式
‑4‑
环戊基环己基)甲氧基)苯基)环己基)
‑
1,2,8
‑
三氟
‑7‑
乙氧基萘52.8g,收率88%,gc纯度99.7%。
[0107][0108]1h nmr(400mhz,cd3cl),δ7.40(d,1h),7.14
–
7.06(m,2h),6.66(d,1h),6.20(d,1h),4.58(t,2h),4.16(d,2h),2.97
‑
2.60(d,2h),2.52
–
2.08(m,6h),1.97
–
1.74(m,3h),1.69
–
1.52(m,12h),1.44
–
0.98(m,9h)。
[0109]
实施例3
[0110]
本实施例提供一种含多氟萘的液晶化合物的制备方法,包括如下步骤:
[0111]
(1)反式
‑4‑
戊基环己基甲醇(ⅳ)的制备:
[0112]
将反式
‑4‑
戊基环己基甲酸19.8g(0.1mol)和102ml甲苯加入500ml三口瓶中,冰浴控温,于0
‑
5℃滴加85.4g氢化二异丁基铝的甲苯溶液(氢化二异丁基铝的质量百分含量为20%,0.12mol),滴加结束后保温搅拌反应2h,于0
‑
5℃条件下滴入90.9ml 20wt%的氢氧化钠水溶液结束反应。然后静置分层,分取有机层,用甲苯萃取水层,将有机层合并,用水洗涤,无水硫酸钠干燥,减压蒸馏脱除溶剂,用37ml乙酸乙酯和148ml正庚烷溶解粗品,过18.4g硅胶柱,收集,浓缩,得反式
‑4‑
戊基环己基甲醇16.9g,收率92%,gc纯度99.6%。
[0113]
(2)1
‑
((反式
‑4‑
戊基环己基)甲氧基)
‑
2,3
‑
二氟苯(ⅲ)的制备:
[0114]
在500ml三口瓶中安装好温度计、机械搅拌和恒压滴液漏斗,加入甲苯150ml、反式
‑4‑
戊基环己基甲醇18.4g(0.1mol)、2,3
‑
二氟苯酚13g(0.1mol)、三苯基膦34.1g(0.13mol),搅拌混合均匀,氮气保护下,冷却至
‑
10℃,滴加偶氮二甲酸二异丙酯26.3g(0.13mol)与131.4ml甲苯的混合溶液,滴加过程温度控制为
‑
10~0℃,滴加结束后升温至25℃,保温2h,保温结束后于25℃加入24.5g水,保温搅拌3h,分液,向水相中加入甲苯萃取,合并有机相,水洗,无水硫酸钠干燥,浓缩,用59ml乙酸乙酯和236ml正庚烷溶解粗品,过29.5g硅胶柱,收集,浓缩,加入正庚烷搅拌结晶0.5h,得1
‑
((反式
‑4‑
戊基环己基)甲氧基)
‑
2,3
‑
二氟苯白色固体26.6g,收率90%,gc纯度99.4%。
[0115]
(3)3
‑
溴
‑
1,2
‑
二氟
‑7‑
丙氧基萘(
ⅸ
)的制备:
[0116]
在500ml三口瓶中安装好温度计、机械搅拌和恒压滴液漏斗,加入甲苯150ml、6
‑
溴
‑
7,8
‑
二氟
‑2‑
萘酚25.9g(0.1mol)、正丙醇6.0g(0.1mol)、三苯基膦34.1g(0.13mol),搅拌混合均匀,氮气保护下,冷却至
‑
10℃,滴加偶氮二甲酸二异丙酯26.3g(0.13mol)与131.4ml甲苯的混合溶液,滴加过程温度控制为
‑
10~0℃,滴加结束后升温至25℃,保温2h,保温结束后于25℃加入24.5g水,保温搅拌3h,分液,向水相中加入甲苯萃取,合并有机相,水洗,无水硫酸钠干燥,浓缩,用60ml乙酸乙酯和240ml正庚烷溶解粗品,过30g硅胶柱,收集,浓缩,加入正庚烷搅拌结晶0.5h,得3
‑
溴
‑
1,2
‑
二氟
‑7‑
丙氧基萘白色固体27.1g,收率90%,gc纯度99.5%。
[0117]
(4)8
‑
(3,4
‑
二氟
‑6‑
丙氧基萘
‑2‑
基)
‑
1,4
‑
二氧杂螺[4.5]癸烷(
ⅶ
)的制备:
[0118]
在1000ml三口瓶中安装好温度计、机械搅拌装置并通入氮气,向体系中加入3
‑
溴
‑
1,2
‑
二氟
‑7‑
丙氧基萘33.1g(0.11mol)和198ml四氢呋喃,搅拌混合均匀,将体系降温至
‑
60℃,然后向体系中保温滴加浓度为1.0mol/l甲基锂的2
‑
甲基四氢呋喃溶液110ml(0.11mol),滴加过程中控温
‑
60℃,滴加结束后保温3h,于
‑
60℃向体系中滴加1,4
‑
环己二酮单乙二醇缩酮15.6g(0.1mol)和50ml四氢呋喃的混合溶液,滴毕,保温反应3h,自然升温至0℃搅拌,之后将体系倒入53ml 10wt%的氯化铵水溶液中进行水解,静置分层,乙酸乙酯萃取,合并有机相,水洗至中性,干燥,浓缩脱除溶剂,将所得固体溶于227ml甲苯中,加入1g对甲基苯磺酸,安装脱水装置,升温至110℃后,回流分水反应0.5h,降至室温,倒入分液漏斗中分液,加水水洗至中性,干燥,浓缩脱干溶剂,向其中加入216ml乙醇,加入1g雷尼镍,于温度50℃、压力0.5mpa条件下进行催化氢化,反应4h后取样分析,加氢反应完毕,过滤,浓缩,用109ml乙酸乙酯和109ml正庚烷溶解粗品,过硅胶柱进行柱层析,收集,浓缩,得8
‑
(3,4
‑
二氟
‑6‑
丙氧基萘
‑2‑
基)
‑
1,4
‑
二氧杂螺[4.5]癸烷31.9g,收率88%,gc纯度99.4%。
[0119]
(5)4
‑
(3,4
‑
二氟
‑6‑
丙氧基萘
‑2‑
基)环己酮(
ⅵ
)的制备:
[0120]
取1000ml三口瓶中安装好温度计、机械搅拌装置,加入8
‑
(3,4
‑
二氟
‑6‑
丙氧基萘
‑2‑
基)
‑
1,4
‑
二氧杂螺[4.5]癸烷36.2g(0.1mol)和200ml甲苯,搅拌混合均匀,加入10wt%甲酸水溶液100ml,于20℃搅拌反应3h,分液,甲苯萃取水相,合并有机相,水洗,浓缩至干,加入正庚烷重结晶,得4
‑
(3,4
‑
二氟
‑6‑
丙氧基萘
‑2‑
基)环己酮29g,收率91%,纯度99.3%。
[0121]
(6)3
‑
(反式
‑4‑
(2,3
‑
二氟
‑4‑
((反式
‑4‑
戊基环己基)甲氧基)苯基)环己基)
‑
1,2
‑
二氟
‑7‑
丙氧基萘(ⅰ)的制备:
[0122]
在1000ml三口瓶中安装好温度计、机械搅拌装置并通入氮气,向体系中加入1
‑
((反式
‑4‑
戊基环己基)甲氧基)
‑
2,3
‑
二氟苯32.6g(0.11mol)和163ml四氢呋喃,搅拌混和均匀,将体系降温至
‑
60℃,然后向体系中保温滴加浓度为1.0mol/l甲基锂的2
‑
甲基四氢呋喃溶液110ml(0.11mol),滴加过程中控温
‑
60℃,滴加结束保温反应4h,于
‑
60℃向体系中滴加4
‑
(3,4
‑
二氟
‑6‑
丙氧基萘
‑2‑
基)环己酮31.8g(0.1mol)和63ml四氢呋喃的混合溶液,滴毕,保温反应4h,自然升温至0℃搅拌,之后将体系倒入100ml 10wt%的乙酸水溶液中水解,静置分层,乙酸乙酯萃取,合并有机相,水洗有机相至中性,干燥,浓缩脱干溶剂后,将所得固体溶于368ml甲苯中,向其中加入1g对甲基苯磺酸,安装脱水装置,升温至110℃后回流分水1h,降温至室温,倒入分液漏斗中,加水水洗至中性,干燥,浓缩脱干溶剂后,向其中加入358ml甲醇,加入5%pd/c催化剂2g,于温度50℃、压力0.5mpa条件下进行催化氢化,反应4h后取样分析,加氢完毕,经过滤、浓缩,用180ml乙酸乙酯和180ml正庚烷溶解粗品,过硅胶柱进行柱层析,收集,浓缩,得3
‑
(反式
‑4‑
(2,3
‑
二氟
‑4‑
((反式
‑4‑
戊基环己基)甲氧基)苯基)环己基)
‑
1,2
‑
二氟
‑7‑
丙氧基萘51.5g,收率86%,gc纯度99.5%。
[0123][0124]1h nmr(400mhz,cd3cl)δ7.70(d,1h),7.47(d,1h),7.20
‑
7.05(m,2h),6.72(d,1h),6.15(t,1h),4.06
‑
3.88(d,4h),2.87(t,1h),2.52(m,1h),2.33
–
2.17(t,6h),1.89
–
1.58(m,8h),1.47
–
1.31(m,10h),1.25
–
1.12(m,7h),0.88(d,3h)。
[0125]
实施例1
‑
3制备的液晶化合物的液晶性能如下:
[0126][0127]
注:光学各向异性(折射率各向异性在25℃下测定
△
n):测定在25℃下,用波长589nm的光,使用阿贝折射仪进行测定
△
n。n
o
对应于寻常光,n
e
对应于非寻常光,δn=n
e
‑
n
o
;
[0128]
介电各向异性(δε):将待测液装入液晶盒中,于25℃温度下,对该液晶盒施加0至30v的电压,在平行于液晶分子长轴方向所测得的平均介电常数为ε
∥
,垂直于液晶分子长轴所测得的平均介电常数为ε
⊥
,介电各向异性δε=ε
∥
‑
ε
⊥
。
[0129]
粘度(γ1):表示旋转粘度(m pa
·
s),测试条件为25
±
0.5℃,20微米平行盒,instec:alct
‑
ng1测试;
[0130]
c.p.表示液晶的清亮点,测试仪器,mettler
‑
toledo
‑
fp system显微热分析仪。
[0131]
实施例4
[0132]
本实施例提供一种含有通式(ⅰ)液晶化合物的液晶组合物,包括如下质量百分含量的组分:
[0133]
[0134]
[0135][0136]
注:表中含有环己基结构的化合物均指其反式结构。
[0137]
其中,“%”表示“质量%”,实施例中测定的特性如下所示:c.p:表示液晶清亮点(℃);
△
n:25℃下测定的折射率各向异性;γ1:25℃下测定的粘度(mpa
·
s)。
[0138]
测试结果:c.p:86.8℃;
△
n:0.103;n
e
:1.605;γ1:89.0mpa
·
s;δε:
‑
4.6;ε
⊥
:8.9。
[0139]
实施例5
[0140]
本实施例提供一种含有通式(ⅰ)液晶化合物的液晶组合物,包括如下质量百分含量的组分:
[0141]
[0142]
[0143][0144]
注:上表中含有环己基结构的化合物均指其反式结构。
[0145]
测试结果:cp:82.5℃;
△
n:0.097;n
e
:1.560;γ1:91mpa
·
s;δε:
‑
4.1;ε
⊥
:8.2
[0146]
实施例6
[0147]
本实施例提供一种含有通式(ⅰ)液晶化合物的液晶组合物,包括如下质量百分含量的组分:
[0148]
[0149][0150][0151]
注:上表中含有环己基结构的化合物均指其反式结构。
[0152]
测试结果:c.p:88.3℃;
△
n:0.105;n
e
:1.615;γ1:96mpa
·
s;δε:
‑
5.1;ε
⊥
:9.5。
[0153]
从以上化合物的液晶性能参数可以看出,本发明液晶化合物具备了作为液晶材料的必要特性,具有适当的光学各向异性,较大负介电各向异性、良好的热稳定性,与其他液
晶化合物混溶性良好,作为显示用液晶材料能够改善液晶组合物的响应速度和驱动电压,可以用作垂直配向方式、ips等液晶显示元件的构成部件。
[0154]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换或改进等,均应包含在本发明的保护范围之内。