1.本发明属于化合物合成及应用技术领域,具体涉及一种5-取代哒嗪-4-胺衍生物、制备方法和用途。
背景技术:
2.恶性肿瘤是当前严重影响人类健康、威胁人类生命的主要疾病之一,己知癌症的种类有100多种,身体的任何部位均可能受到癌症的侵袭,因此,攻克癌症被列为研究的首要任务,开发能够有效治疗肿瘤、毒副作用小、耐药性低的药物的研究课题,已经成为了科学家们急需解决的重要的问题。
3.哒嗪是一类具有广泛生物活性的杂环化合物,在药物研究领域具有广泛的应用。自从第一例天然存在的具有杀菌活性的哒嗪衍生物哒嗪霉菌素(pyridazomycin)被报道后,哒嗪类化合物的研究引起了人们广泛的兴趣、。目前已经有多种哒嗪类药物被开发上市,如治疗心理疾病的药物米那普林(minaprine)、抗菌药物长效磺胺(sulfapiridazin)、降压药物双肼苯哒嗪(nepressol)、广谱抗生素类药物磺胺氯哒嗪(sulfachloropyridazine)、促进血液循环药物甲硫阿美铵(ameziniummetilsulfate)等。由于哒嗪类化合物所表现出的广泛的药理和生理活性,合成并开发结构新颖的哒嗪衍生物具有重要意义。近年来,围绕哒嗪衍生物的合成及其抗肿瘤活性研究已经成为药物研究领域的热点课题之一。然而,目前已经被报道合成的取代哒嗪类化合物主要以哒嗪酮类以及3,6-位取代哒嗪衍生物为主,根据文献调研发现,哒嗪酮及3,6-位取代哒嗪衍生物主要应用于农药领域作为杀虫剂、杀菌剂、除草剂、植物生长调节剂等,在抗肿瘤药物领域目前尚处于研究阶段。而关于4,5-位取代的哒嗪衍生物的合成及抗肿瘤活性研究则相对报道较少。
技术实现要素:
4.本发明公开了一种5-取代哒嗪-4-胺衍生物,毒性低,且该衍生物对肿瘤细胞具有优异的抑制活性,能作为抗肿瘤性疾病的药物广泛应用,同时为哒嗪类抗肿瘤药物的研究开发提供一种新的思路。
5.一种5-取代哒嗪-4-胺衍生物在制备抗肿瘤药物中的用途。
6.进一步的,所述肿瘤为肺癌、前列腺癌、乳腺癌、肝癌或宫颈癌。
7.进一步的,所述5-取代哒嗪-4-胺衍生物的结构式如式(ⅰ)所示:
[0008][0009]
其中,r是烯基或芳基。
[0010]
进一步的,所述r是烯基时,烯基为烯丙基或环戊烯基;所述r是芳基时,芳基为苯基、取代苯基、取代吡啶基、萘基、吲哚基或喹啉基。
[0011]
一种5-取代哒嗪-4-胺衍生物,结构式如式(ⅰ)所示:
[0012][0013]
其中,r是烯基或芳基。
[0014]
进一步的,所述烯基为烯丙基或环戊烯基;所述芳基为苯基、取代苯基、取代吡啶基、萘基、吲哚基或喹啉基。
[0015]
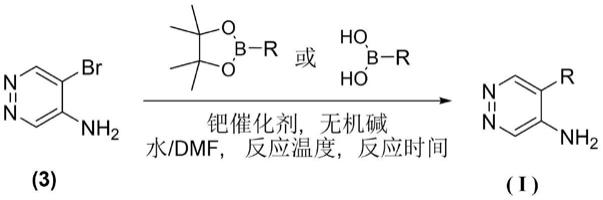
一种5-取代哒嗪-4-胺衍生物的制备方法,包括以下步骤:
[0016]
1)在氮气保护下,将5-溴哒嗪-4-胺、n,n-二甲基甲酰胺和水中,得到混合液a;
[0017]
2)向步骤1)的混合液中加入化合物a、无机碱和钯催化剂,在80℃~120下搅拌1~5小时;得到混合液b;
[0018]
3)将混合液b加水稀释,用乙酸乙酯萃取,取有机相经洗涤、干燥,减压浓缩得到粗产品;
[0019]
4)得到的粗产品经柱层析分离纯化,得到目标产物。
[0020]
进一步的,所述步骤1)中,5-溴哒嗪-4-胺与n,n-二甲基甲酰胺的质量体积比为1000~3000mg:20~50ml;所述n,n-二甲基甲酰胺和水的体积比为1~4:1~10。
[0021]
进一步的,所述步骤2)中,化合物a与5-溴哒嗪-4-胺的摩尔质量比为1.0:1~2.0:1。
[0022]
进一步的,所述步骤2)中,化合物a为r是烯基或芳基;所述无机碱为碳酸钠、碳酸钾或碳酸铯;所述钯催化剂为pd(dppf)cl2、pd(pph3)4、pd(oac)2/pph3或pd(dba)2。
[0023]
本发明的有益效果是:
[0024]
1、本发明提供的5-取代哒嗪-4-胺衍生物,这类化合物对人非小细胞肺癌a549细胞、人前列腺癌pc3细胞、人乳腺癌mcf-7细胞、人肝癌细胞hepg2、人宫颈癌hela细胞均具有较明显的抑制活性,尤其是对人肝癌hepg2细胞具有优异的抑制活性,而且毒性较低,可作为抗肿瘤性疾病的药物;同时为哒嗪类抗肿瘤药物的研究开发提供一种新的思路。
[0025]
2、本发明提供的5-取代哒嗪-4-胺衍生物,制备方法简单,合成条件温和,产率高,毒性低,安全性高,可广泛应用于药物中。
附图说明
[0026]
图1是实施例1制备的式(ia)所示化合物的1h-nmr图;
[0027]
图2是实施例2制备的式(ib)所示化合物的1h-nmr图;
[0028]
图3是实施例3制备的式(ic)所示化合物的1h-nmr图;
[0029]
图4是实施例4制备的式(id)所示化合物的1h-nmr图;
[0030]
图5是实施例5制备的式(ie)所示化合物的1h-nmr图;
[0031]
图6是实施例6制备的式(if)所示化合物的1h-nmr图;
[0032]
图7是实施例7制备的式(ig)所示化合物的1h-nmr图;
[0033]
图8是实施例8制备的式(ih)所示化合物的1h-nmr图;
[0034]
图9是实施例9制备的式(ii)所示化合物的1h-nmr图;
[0035]
图10是实施例10制备的式(ij)所示化合物的1h-nmr图;
[0036]
图11是实施例11制备的式(ik)所示化合物的1h-nmr图。
具体实施方式
[0037]
现结合附图以及实施例对本发明做详细的说明。
[0038]
本发明提供的5-取代哒嗪-4-胺衍生物,且对肿瘤细胞具有优异的抑制活性,能作为抗肿瘤性疾病的药物广泛应用,同时为哒嗪类抗肿瘤药物的开发应用提供一种新的思路。
[0039]
进一步的,5-取代哒嗪-4-胺衍生物对肺癌、前列腺癌、乳腺癌、肝癌或宫颈癌具有优异的抑制活性。
[0040]
本发明提供的5-取代哒嗪-4-胺衍生物,结构式如式(ⅰ)所示:
[0041][0042]
其中,r是烯基或芳基。
[0043]
进一步的,r是烯基时,烯基为烯丙基或环戊烯基。
[0044]
进一步的,r是芳基时,芳基为苯基、取代苯基、取代吡啶基、萘基、吲哚基或喹啉基。
[0045]
本发明提供的5-取代哒嗪-4-胺衍生物的制备路线是:
[0046][0047]
具体的制备方法,包括以下步骤:
[0048]
1)在氮气保护下,将5-溴哒嗪-4-胺n,n-二甲基甲酰胺dmf和水中,得到混合液a;
[0049]
2)向步骤1)的混合液中加入化合物a、无机碱和钯催化剂,在80℃~120下搅拌1~5小时;得到混合液b;
[0050]
3)将混合液b加水稀释,用乙酸乙酯萃取,取有机相经洗涤、干燥,减压浓缩得到粗产品;
[0051]
4)得到的粗产品经柱层析分离纯化,得到目标产物。
[0052]
进一步的,所述步骤1)中,5-溴哒嗪-4-胺与n,n-二甲基甲酰胺的质量体积比为
1000~3000mg:20~50ml;所述n,n-二甲基甲酰胺和水的体积比为1~4:1~10。
[0053]
进一步的,所述步骤2)中,化合物a与5-溴哒嗪-4-胺的摩尔质量比为1.0:1~2.0:1。
[0054]
本发明提供的步骤2)中,化合物a为无机碱为碳酸钠、碳酸钾或碳酸铯;钯催化剂为pd(dppf)cl2、pd(pph3)4、pd(oac)2/pph3或pd(dba)2。
[0055]
下面以具体的几个具体的5-取代哒嗪-4-胺衍生物为例,说明本发明衍生物的制备以及应用。
[0056]
实施例1 5-(1-丙烯-2-基)吡啶哒嗪-4-胺的合成,其结构式如(ia)所示,
[0057][0058]
本实施例结构式如(ia)所示的化合物,其制备方法是:
[0059]
1)在氮气保护下,将5-溴哒嗪-4-胺(3)(1.7g,10.0mmol)溶解于n,n-二甲基甲酰胺(30ml)和水(5ml)的混合溶液中,得到混合液a;
[0060]
2)然后向混合液a中加入异丙烯基硼酸频哪醇酯(2.4g,14.0mmol)、碳酸钠(2.1g,20.0mmol)和pd(dppf)cl2(366mg,0.50mmol),混合物在100℃下搅拌2小时;得到混合液b;
[0061]
3)然后混合液b加水(60ml)稀释,用乙酸乙酯(3
×
100ml)萃取,有机相经饱和食盐水洗涤、无水硫酸钠干燥,然后减压浓缩,得到粗品;
[0062]
4)粗品经柱层析分离纯化,得到黄色固体。分离纯化时采用的洗脱剂为二氯甲烷-甲醇混合溶剂(体积比20:1)。
[0063]
本实施例中,得到的黄色固体的收率58%。黄色固体即为5-(1-丙烯-2-基)吡啶哒嗪-4-胺化合物,进一步对进行核磁共振,得到氢谱,如图1所示。
[0064]
从图1可知,1h nmr(400mhz,dmso-d6)δ8.54(s,1h),8.37(s,1h),6.22(br,2h),5.37~5.31(m,1h),5.14(s,1h),2.01(d,j=0.6hz,3h)。
[0065]
实施例2 5-(环戊-1-烯-1-基)哒嗪-4-胺的合成,其结构式如(ib)所示,
[0066][0067]
本实施例中,结构式如(ib)所示的化合物的制备方法,包括:
[0068]
1)在氮气保护下,将5-溴哒嗪-4-胺(3)(1.7g,10.0mmol)溶解于n,n-二甲基甲酰胺(30ml)和水(5ml)的混合溶液中,得到混合液a;
[0069]
2)然后向混合液a中加入1-环戊烯硼酸频哪醇酯(2.7g,14.0mmol)、碳酸钠(2.1g,20.0mmol)和pd(dppf)cl2(366mg,0.50mmol),混合物在100℃下搅拌2小时,得到混合液b;
[0070]
3)然后向混合液b中加水(60ml)稀释,用乙酸乙酯(3
×
100ml)萃取,有机相经饱和食盐水洗涤、无水硫酸钠干燥,然后减压浓缩,得到粗品;
[0071]
4)粗品经柱层析分离纯化,得到黄色固体。分离纯化时采用的洗脱剂为二氯甲烷-甲醇混合溶剂(体积比20:1)。
[0072]
本实施例中,得到的黄色固体的收率64%。黄色固体即为5-(环戊-1-烯-1-基)哒嗪-4-胺化合物,进一步对进行核磁共振,得到氢谱,如图2所示。
[0073]
参见图2,1h nmr(400mhz,dmso-d6)δ8.56(s,1h),8.43(s,1h),6.31(br,2h),6.26(s,1h),2.70~2.66(m,1h),2.56~2.44(m,3h),1.90(dd,j=14.9,7.5hz,2h),具体见附图2。
[0074]
实施例3 5-苯基吡啶-4-胺的合成,其结构式如(ic)所示,
[0075][0076]
本实施例中,结构式如(ic)所示的化合物的制备方法,包括:
[0077]
1)在氮气保护下,将5-溴哒嗪-4-胺(3)(1.7g,10.0mmol)溶解于n,n-二甲基甲酰胺(30ml)和水(5ml)的混合溶液中,得到混合液a;
[0078]
2)然后向混合液a中加入苯硼酸(1.7g,14.0mmol)、碳酸钠(2.1g,20.0mmol)和pd(dppf)cl2(366mg,0.50mmol),混合物在100℃下搅拌2小时,得到混合液b;
[0079]
3)然后混合液b中加水(60ml)稀释,用乙酸乙酯(3
×
100ml)萃取,有机相经饱和食盐水洗涤、无水硫酸钠干燥,然后减压浓缩,得到粗品;
[0080]
4)粗品经柱层析分离纯化,得到白色固体。分离纯化时采用的洗脱剂为二氯甲烷-甲醇混合溶剂(体积比20:1)。
[0081]
本实施例中,得到的白色固体的收率84%。白色固体即为5-苯基吡啶-4-胺化合物,进一步对进行核磁共振,得到氢谱,如图3所示。
[0082]
参见图3,1h nmr(400mhz,dmso-d6)δ8.64(s,1h),8.45(s,1h),7.54~7.42(m,5h),6.28(br,2h),具体见附图3。
[0083]
实施例4 5-(3-氟-5-甲基苯基)哒嗪-4-胺的合成,其结构式如(id)所示,
[0084][0085]
本实施例中,结构式如(id)所示的化合物的制备方法,包括:
[0086]
1)在氮气保护下,将5-溴哒嗪-4-胺(3)(1.7g,10.0mmol)溶解于n,n-二甲基甲酰胺(30ml)和水(5ml)的混合溶液中,得到混合液a;
[0087]
2)然后混合液a中加入3-氟-5-甲基苯硼酸(2.2g,14.0mmol)、碳酸钠(2.1g,20.0mmol)和pd(dppf)cl2(366mg,0.50mmol),混合物在100℃下搅拌2小时,得到混合液b;
[0088]
3)然后向混合液b中加水(60ml)稀释,用乙酸乙酯(3
×
100ml)萃取,有机相经饱和食盐水洗涤、无水硫酸钠干燥,然后减压浓缩,得到粗品;
[0089]
4)粗品经柱层析分离纯化,得到淡黄色固体。分离纯化时采用的洗脱剂为二氯甲烷-甲醇混合溶剂(体积比20:1)。
[0090]
本实施例中,得到的淡黄色固体的收率82%。淡黄色固体即为5-(3-氟-5-甲基苯基)哒嗪-4-胺化合物,进一步对进行核磁共振,得到氢谱,如图4所示。
[0091]
参见图4,1h nmr(400mhz,dmso-d6)δ8.64(s,1h),8.45(s,1h),7.20~7.02(m,3h),6.39(br,2h),2.38(s,3h),具体见附图4。
[0092]
实施例5 3-(5-氨基吡嗪-4-基)苯甲腈的合成,结构式如(ie)所示;
[0093][0094]
本实施例中,结构式如(ie)所示的化合物的制备方法,包括:
[0095]
1)在氮气保护下,将5-溴哒嗪-4-胺(3)(1.7g,10.0mmol)溶解于n,n-二甲基甲酰胺(30ml)和水(5ml)的混合溶液中,得到混合液a;
[0096]
2)然后向混合液a中加入3-氰基苯硼酸(2.1g,14.0mmol)、碳酸钠(2.1g,20.0mmol)和pd(dppf)cl2(366mg,0.50mmol),混合物在100℃下搅拌2小时,得到混合液b;
[0097]
3)然后向混合液b中加水(60ml)稀释,用乙酸乙酯(3
×
100ml)萃取,有机相经饱和食盐水洗涤、无水硫酸钠干燥,然后减压浓缩,得到粗品;
[0098]
4)粗品经柱层析分离纯化,得到白色固体。分离纯化时采用的洗脱剂为二氯甲烷-甲醇混合溶剂(体积比20:1)。
[0099]
本实施例中,得到的白色固体的收率88%。白色固体即为3-(5-氨基吡嗪-4-基)苯甲腈化合物,进一步对进行核磁共振,得到氢谱,如图5所示。
[0100]
参见图5,1h nmr(400mhz,dmso-d6)δ8.65(s,1h),8.48(s,1h),7.95(s,1h),7.90(d,j=7.7hz,1h),7.81(d,j=7.6hz,1h),7.70(t,j=7.7hz,1h),6.51(br,2h),具体见附图5。
[0101]
实施例6 3-(5-氨基吡啶-4-基)-5-氟苯腈的合成,其结构式如(if)所示,
[0102][0103]
本实施例中,结构式如(if)所示的化合物的制备方法,包括:
[0104]
1)在氮气保护下,将5-溴哒嗪-4-胺(3)(1.7g,10.0mmol)溶解于n,n-二甲基甲酰胺(30ml)和水(5ml)的混合溶液中,得到混合液a;
[0105]
2)然后向混合液a中加入3-氟-5-氰基苯硼酸(2.3g,14.0mmol)、碳酸钠(2.1g,20.0mmol)和pd(dppf)cl2(366mg,0.50mmol),混合物在100℃下搅拌2小时,得到混合液b;
[0106]
3)然后向混合液b中加水(60ml)稀释,用乙酸乙酯(3
×
100ml)萃取,有机相经饱和食盐水洗涤、无水硫酸钠干燥,然后减压浓缩,得到粗品;
[0107]
4)粗品经柱层析分离纯化,得到棕色固体。分离纯化时采用的洗脱剂为二氯甲烷-甲醇混合溶剂(体积比20:1)。
[0108]
本实施例中,得到的棕色固体的收率81%。棕色固体即为3-(5-氨基吡啶-4-基)-5-氟苯腈化合物,进一步对进行核磁共振,得到氢谱,如图6所示。
[0109]
参见图6,1h nmr(400mhz,dmso-d6)δ8.66(s,1h),8.50(s,1h),7.92(d,j=7.9hz,1h),7.83(s,1h),7.75(d,j=9.6hz,1h),6.61(br,2h),具体见附图6。
[0110]
实施例7 5-(2-氟-5-甲氧基苯基)哒嗪-4-胺的合成,其结构式如(ig)所示,
[0111][0112]
本实施例中,结构式如(ig)所示的化合物的制备方法,包括:
[0113]
1)在氮气保护下,将5-溴哒嗪-4-胺(3)(1.7g,10.0mmol)溶解于n,n-二甲基甲酰胺(30ml)和水(5ml)的混合溶液中,得到混合液a;
[0114]
2)然后向混合液a中加入(2-氟-5-甲氧基吡啶-3-基)苯硼酸(2.4g,14.0mmol)、碳酸钠(2.1g,20.0mmol)和pd(dppf)cl2(366mg,0.50mmol),混合物在100℃下搅拌2小时,得到混合液b;
[0115]
3)然后向混合液b中加水(60ml)稀释,用乙酸乙酯(3
×
100ml)萃取,有机相经饱和食盐水洗涤、无水硫酸钠干燥,然后减压浓缩,得到粗品;
[0116]
4)粗品经柱层析分离纯化,得到白色固体。分离纯化时采用的洗脱剂为二氯甲烷-甲醇混合溶剂(体积比20:1)。
[0117]
本实施例中,得到的白色固体的收率74%。白色固体即为5-(2-氟-5-甲氧基苯基)哒嗪-4-胺化合物,进一步对进行核磁共振,得到氢谱,如图7所示。
[0118]
参见图7,1hnmr(400mhz,dmso-d6)δ8.64(s,1h),8.41(s,1h),7.27(t,j=9.3hz,1h),7.06
–
7.00(m,1h),6.95(dd,j=5.8,3.2hz,1h),6.34(br,2h),3.78(s,3h)。
[0119]
实施例8 5-(萘-2-基)哒嗪-4-胺的合成,其结构式如(ih)所示,
[0120][0121]
本实施例中,结构式如(ih)所示的化合物的制备方法,包括:
[0122]
1)在氮气保护下,将5-溴哒嗪-4-胺(3)(1.7g,10.0mmol)溶解于n,n-二甲基甲酰胺(30ml)和水(5ml)的混合溶液中,得到混合液a;
[0123]
2)然后向混合液a中加入2-萘硼酸(2.4g,14.0mmol)、碳酸钠(2.1g,20.0mmol)和pd(dppf)cl2(366mg,0.50mmol),混合物在100℃下搅拌2小时,得到混合液b;
[0124]
3)然后向混合液b中加水(60ml)稀释,用乙酸乙酯(3
×
100ml)萃取,有机相经饱和食盐水洗涤、无水硫酸钠干燥,然后减压浓缩,得到粗品;
[0125]
4)粗品经柱层析分离纯化,得到白色固体。分离纯化时采用的洗脱剂为二氯甲烷-甲醇混合溶剂(体积比20:1)。
[0126]
本实施例中,得到的白色固体的收率82%。白色固体即为5-(萘-2-基)哒嗪-4-胺化合物,进一步对进行核磁共振,得到氢谱,如图8所示。
[0127]
参见图8,1h nmr(400mhz,dmso-d6)δ8.68(s,1h),8.57(s,1h),8.05(d,j=7.5hz,2h),7.99(dd,j=6.2,3.2hz,2h),7.59(ddd,j=9.6,7.4,2.4hz,3h),6.39(br,2h)。
[0128]
实施例9 5-(1h-吲哚-6-基)哒嗪-4-胺的合成,其结构式如(ii)所示,
[0129][0130]
本实施例中,结构式如(ii)所示的化合物的制备方法,包括:
[0131]
1)在氮气保护下,将5-溴哒嗪-4-胺(3)(1.7g,10.0mmol)溶解于n,n-二甲基甲酰胺(30ml)和水(5ml)的混合溶液中,得到混合液a;
[0132]
2)然后向混合液a中加入6-吲哚硼酸(2.3g,14.0mmol)、碳酸钠(2.1g,20.0mmol)和pd(dppf)cl2(366mg,0.50mmol),混合物在100℃下搅拌2小时,得到混合液b;
[0133]
3)然后向混合液b中加水(60ml)稀释,用乙酸乙酯(3
×
100ml)萃取,有机相经饱和食盐水洗涤、无水硫酸钠干燥,然后减压浓缩,得到粗品;
[0134]
4)粗品经柱层析分离纯化,得到棕色固体。分离纯化时采用的洗脱剂为二氯甲烷-甲醇混合溶剂(体积比15:1)。
[0135]
本实施例中,得到的棕色固体的收率77%。棕色固体即为5-(3-氟-5-甲基苯基)哒嗪-4-胺化合物,进一步对进行核磁共振,得到氢谱,如图9所示。
[0136]
参见图9,1h nmr(400mhz,dmso-d6)δ11.28(s,1h),8.64(s,1h),8.49(s,1h),7.67(d,j=8.1hz,1h),7.51(s,1h),7.44(t,j=2.7hz,1h),7.08(d,j=8.1hz,1h),6.50(s,1h),6.21(br,2h)。
[0137]
实施例10 5-(2-甲氧基喹啉-3-基)哒嗪-4-胺的合成,其结构式如(ij)所示,
[0138][0139]
本实施例中,结构式如(ij)所示的化合物的制备方法,包括:
[0140]
1)在氮气保护下,将5-溴哒嗪-4-胺(3)(1.7g,10.0mmol)溶解于n,n-二甲基甲酰胺(30ml)和水(5ml)的混合溶液中,得到混合液a;
[0141]
2)然后向混合液a中加入(2-甲氧基喹啉-3-基)硼酸(2.8g,14.0mmol)、碳酸钠(2.1g,20.0mmol)和pd(dppf)cl2(366mg,0.50mmol),混合物在100℃下搅拌2小时,得到混
合液b;
[0142]
3)然后向混合液b中加水(60ml)稀释,用乙酸乙酯(3
×
100ml)萃取,有机相经饱和食盐水洗涤、无水硫酸钠干燥,然后减压浓缩,得到粗品;
[0143]
4)粗品经柱层析分离纯化,得到白色固体。分离纯化时采用的洗脱剂为二氯甲烷-甲醇混合溶剂(体积比15:1)。
[0144]
本实施例中,得到的白色固体的收率70%。白色固体即为5-(3-氟-5-甲基苯基)哒嗪-4-胺化合物,进一步对进行核磁共振,得到氢谱,如图10所示。
[0145]
参见图10,1hnmr(400mhz,dmso-d6)δ8.63(s,1h),8.44(s,1h),8.24(s,1h),7.94(d,j=8.0hz,1h),7.85(d,j=8.3hz,1h),7.72(t,j=7.6hz,1h),7.48(t,j=7.5hz,1h),6.35(br,2h),3.98(s,3h)。
[0146]
实施例11 5-[4-(4-乙基哌嗪-1-甲基)苯基]哒嗪-4-胺的合成,其结构式如(ik)所示,
[0147][0148]
本实施例中,结构式如(ik)所示的化合物的制备方法,包括:
[0149]
1)在氮气保护下,将5-溴哒嗪-4-胺(3)(1.7g,10.0mmol)溶解于n,n-二甲基甲酰胺(30ml)和水(5ml)的混合溶液中,得到混合液a;
[0150]
2)然后在混合液a中加入4-[(4-乙基哌嗪-1-基)甲基]苯硼酸(3.5g,14.0mmol)、碳酸钠(2.1g,20.0mmol)和pd(dppf)cl2(366mg,0.50mmol),混合物在100℃下搅拌2小时,得到混合液b;
[0151]
3)然后向混合液b中加水(60ml)稀释,用乙酸乙酯(3
×
100ml)萃取,有机相经饱和食盐水洗涤、无水硫酸钠干燥,然后减压浓缩,得到粗品;
[0152]
4)粗品经柱层析分离纯化,得到白色固体。分离纯化时采用的洗脱剂为二氯甲烷-甲醇混合溶剂(体积比10:1)。
[0153]
本实施例中,得到的白色固体的收率68%。白色固体即为5-[4-(4-乙基哌嗪-1-甲基)苯基]哒嗪-4-胺化合物,进一步对进行核磁共振,得到氢谱,如图11所示。
[0154]
参见图11,1h nmr(400mhz,dmso-d6)δ8.63(s,1h),8.45(s,1h),7.48
–
7.38(m,4h),6.27(br,2h),3.48
–
3.33(m,4h),2.39-2.35(m,8h),1.00(t,j=7.1hz,3h)。
[0155]
实施例12式(ⅰ)所示化合物的体外抗肿瘤活性
[0156]
选取人非小细胞肺癌a549细胞、前列腺癌细胞pc-3、人乳腺癌细胞mcf-7、hepg2肝癌细胞、人宫颈癌hela细胞为测试细胞株。以5-氟尿嘧啶(5-fu)为阳性对照药,采用mts法对式(ia~ik)所示化合物进行体外抗肿瘤活性评价。
[0157]
具体的过程是,分别将样品(对照药以及ia~ik所示化合物)用dmso溶解后,然后用dmem培养基稀释成50、20、10、5、1、0.5、0.1、0.01μmol/l不同浓度。取对数生长期的测试细胞株悬浮于含10%胎牛血清的无酚红dmem培养基中,铺至96孔细胞培养板中,每孔加入100μl(4~10)
×
104个/ml细胞悬液。待细胞完全贴壁后,弃去原培养液,加入100μl的含有
测试药物的培养液培养3天后,每孔加入30μl 5mg/ml mtt,置于37℃、5%co2培养箱中继续孵育4h,然后每孔加入100μl二甲亚砜(dmso)溶解。使用酶标仪在490nm波长测定每孔的吸光度(od)值,计算被测药物对肿瘤细胞的抑制率(%),采用非线性回归模型绘制s型剂量效应曲线,用originpro软件计算出半数抑制浓度(ic
50
)值,结果见表1。
[0158]
细胞抑制率(%)=(正常od值-加药od值)/正常od值
×
100%
[0159]
表1化合物ia-ik的抗细胞增值活性(ic
50
,μm)a[0160][0161]
表1的抗细胞增值活性测试结果表明,测试样品式(ia-ik)对于5种肿瘤细胞均具有明显的抑制活性,尤其是对于hepg2肝癌细胞具有较好抑制活性,ic
50
=8.4μm,说明该结构具有潜在抗肿瘤活性,可用作抗肿瘤活性先导化合物。
[0162]
此外,所有目标化合物对健康的lo2肝细胞无毒性,而5-氟尿嘧啶表现出相当大的毒性(表2)。
[0163]
表2和ia-ik的体外治疗指数(ivti)a[0164][0165]
比较它们对hepg2细胞的作用,我们的化合物ia-ik在体外治疗指数(ivti,表2)比5-fu大得多,说明我们的化合物的生物安全性更高。