1.本发明属于基因工程领域,涉及利用基因工程方法改良真菌性状。
背景技术:
2.昆虫生防真菌是一类重要的昆虫生防微生物,菌种资源丰富,是自然界控制害虫群体的主要因素之一(roberts et al.,2004,adv.appl.microbiol.,54:1
–
70)。与细菌和病毒类昆虫病原微生物通过消化道侵染方式不同,昆虫病原真菌是唯一直接穿透体壁侵染昆虫的微生物,对于控制将口针直接插入植物韧皮部吸食汁液的刺吸式口器害虫如蚜虫、叶蝉、飞虱等具有独到优势。因此,开发和应用真菌杀虫剂受到国内外的广泛关注。其中,多种昆虫生防真菌如以球孢白僵菌(beauveria bassiana)、罗伯茨绿僵菌(metarhizium robertsii)、蝗绿僵菌(m.acridum)等为活性成分开发的真菌生防制剂广泛用于农、林以及卫生害虫的生物防治(fan et al.,2012;yang et al.,2014)。开发和应用真菌杀虫剂受到国内外的广泛关注,如以球孢白僵菌为活性成分在全球注册登记的生防制剂已超过170余种(de faria et al.,2007,biol control,43:237-256)。
3.昆虫病原真菌的杀虫方式主要是通过活体成分穿透靶标昆虫体壁,引起昆虫侵染致病并致死昆虫。真菌侵染昆虫过程涉及侵染单元分生孢子或芽生孢子附着于昆虫体壁,萌发产生芽管并在昆虫体表进行水平生长,生长菌丝在昆虫的节间膜等特殊部分分化形成侵染结构或附着胞,然后由侵染结构分化形成侵染菌丝,在机械压力和分泌产生的一系列的水解酶如酯酶、几丁酶、蛋白酶的作用等的协同作用下穿透昆虫体壁。菌丝一旦穿入体壁进入血腔,菌丝以芽殖方式分化为芽生孢子,称之为虫菌体,有助于真菌逃避昆虫天然免疫(包括细胞免疫和体液免疫)反应。同时,真菌一方面通过特化的细胞结构“伪装”病原相关分子模式,逃避昆虫免疫识别,并分泌代谢产物如卵胞菌素(oosporein)(feng et al.,2015,proc natl acad sci usa 112:11365
–
11370)、破坏素(destruxins)(wang et al.,2012,proc natl acad sci usa 109:1287
–
1292)等和效应分子(cen et al.,2017,plos pathog 13(9):e1006604)抑制宿主免疫反应;另一方面产生一系列的水解酶降解昆虫组织,掠夺昆虫营养促进病原菌增殖,引起昆虫致病死亡,菌体充满虫体形成“僵虫”。在合适的温、湿度条件下,菌体又从致死的僵虫体内穿出体壁生长和产孢进行扩散,引起新的侵染循环,实现持续控制害虫的目的。然而,真菌侵染致病的进程决定了其“击倒”靶标速度较慢,在一定程度上影响生防制剂的应用范围。解析生防真菌侵染致病的分子机制,挖掘提升真菌侵染效果的功能基因,是改良和提升生防真菌应用效果的重要途径。
4.昆虫生防真菌生长及繁殖速度、产孢能力等性状与真菌制剂的生产密切相关,是真菌生防制剂的生产重要参数。目前,生产上提高产孢量的方法主要是通过培养基及培养条件优化,促进生长和产孢,而利用基因操作促进真菌生产和产孢,鲜有报道。
5.β-葡聚糖酶是一类能够水解以β-糖苷键连接的葡聚糖的酶系,广泛分布在植物、细菌、真菌和病毒等组织中(doxey et al.,2007,mol biol evol 24:1045-55)。研究发现,β-葡聚糖酶参与真菌细胞壁代谢、发育分化及侵染致病等生物学过程。烟曲霉
(aspergillus fumigatus)4个β-1,3-葡聚糖酶编码基因(eng2-5)在分生孢子休眠和萌发过程中均有表达,破坏eng2,3,4或5均导致产孢缺陷,分生孢子不能正常分离,表明内切β-1,3-葡聚糖酶对于丝状真菌中分生孢子细胞壁的正确组装和分生孢子的分离是必需的(mouyna et al.,2016,cell microbiol,18(9):1285-1293)。破坏酿酒酵母(saccharomyces cerevisiae)的β-葡聚糖酶基因eng1导致细胞出现分离缺陷。敲除粟酒裂殖酵母(schizosaccharomyces pombe)中eng1同源基因后出现相似的性状,由于母细胞与子细胞之间的初生壁中β-1,3-葡聚糖无法降解而导致不能正常分离(baladr
ó
n et al.,2002,eukaryot cell 1:774-786;martin-cuadrado,2003,j cell sci 116:1689-1698)。葡聚糖酶在植物病原菌致病过程中起着很重的作用。大豆疫霉(phytophthora sojae)在侵染大豆的过程中,gh12家族的葡聚糖酶psxeg1发挥致病毒力因子和病原相关模式分子双重角色,敲除psxeg1的病原菌毒力显著降低(ma,et al.,2015,plant cell 27:2057)。敲除灰葡萄孢菌(botrytis cinerea)的木葡聚糖酶(xynl1a)基因显著降低病原菌对葡萄和番茄的致病力等(n
é
lida brito et al.,2006,mol plant microbe interact 19:25-32)。由此表明,β-葡聚糖酶蛋白与真菌生长发育和侵染宿主过程密切相关。
6.在解析重要昆虫病原真菌球孢白僵菌(b.bassiana)侵入昆虫血腔后增殖细胞(虫菌体)表面蛋白时,发现β-葡聚糖酶eng1仅存在于虫菌体表面蛋白谱,而不存在于体外培养的真菌细胞表面蛋白谱。采用基因表达、分泌特性、基因敲除及过表达以及酶学功能分析发现,bbeng1基因表达受昆虫营养诱导,并可分泌至胞外结合和降解昆虫体壁及血腔营养。基因敲除导致菌株毒力下降,而过表达基因促进菌株生长和分生孢子产生,毒力显著增强,提高杀虫效果。此外,研究发现,bbeng1分布于细胞壁和细胞表面,分布于细胞壁,参与细胞壁的代谢更新,参与菌体增殖;作为表面蛋白,“隐蔽”病原相关分子模式如β-1,3-葡聚糖等,有助于病原菌逃避昆虫免疫识别。将bbeng1基因导入罗伯茨绿僵菌(m.robertsii)和蝗绿僵菌(m.acridum)中也明显促进菌株生长和产孢,毒力显著增强。此外,分别将罗伯茨绿僵菌和蝗绿僵菌的同源基因mreng1和maeng1分别导入各自菌株,也明显促进了菌株生长和产孢能力,显著增强了菌株毒力。
技术实现要素:
7.本发明的一个目的是提供一种促进昆虫生防真菌球孢白僵菌、罗伯茨绿僵菌和蝗绿绿僵菌生长和分生孢子产量及提高毒力的方法。
8.本发明的另一个目的是提供一种通过基因工程方法构建的球孢白僵菌、罗伯茨绿僵菌和蝗绿僵菌工程菌株。
9.本发明的再一个目的是提供一类真菌杀虫剂。
10.本发明也提供bbeng1、mreng1和maengl基因在制备真菌杀虫剂中的用途。其中,bbeng1(bba_04753)定位于球孢白僵菌基因组序列nw_007930846.1区域(359177bp-372868bp)的365011bp-366984bp之间(https://www.ncbi.nlm.nih.gov/gene/?term=bba_04753),mreng1(maa_09026)定位于罗伯茨绿僵菌基因组序列nw_011942204.1区域(26922bp-44989bp)的37770bp-39256bp之间(https://www.ncbi.nlm.nih.gov/gene/?term=maa_09026),maeng1(mac_06610)定位于蝗绿僵菌基因组序列nw_006916732.1区域(139846bp-158861bp)的148024bp-149543bp之间(https://www.ncbi.nlm.nih.gov/
gene/?term=mac_06610)。
11.根据本发明的一方面,一种提高昆虫生防真菌生长、产孢能力和提高毒力的方法,其特征在于通过构建超量表达昆虫生防真菌分泌性葡聚糖酶基因eng1的工程菌株,获得具有促进生长、提高分生孢子产量和毒力的昆虫生防真菌。
12.本发明所述的昆虫生防真菌选自球孢白僵菌(beauveria bassiana),罗伯茨绿僵菌(metarhizium robertsii)和蝗绿僵菌(m.acridum);所述昆虫生防真菌分泌性葡聚糖酶基因eng1选自下述一种:来源于球孢白僵菌的分泌性葡聚糖酶基因bbeng1,来源于罗伯茨绿僵菌的bbeng1同源基因mreng1和来源于蝗绿僵菌的bbeng1同源基因maeng1。
13.本发明所述的方法中,将所述昆虫生防真菌分泌性葡聚糖酶基因eng1置于真菌组成型启动子控制下,构建超量表达载体;所述真菌基因组成型启动子选自pgpda和pb3。
14.本发明优选的方法是通过转入组成型启动子pgpda控制目的基因bbeng1至球孢白僵菌野生型菌株,rt-qpcr筛选验证获得bbeng1超量表达工程菌株,所述工程菌株中bbeng1结构域编码区高水平转录。
15.具体的,超量表达bbeng1基因编码的gh16结构域优选的是以真菌基因组成型启动子pgpda(来源于构巢曲霉3-磷酸甘油醛脱氢酶基因启动子)为实现超量表达的启动子,使用以草甘膦抗性基因bar为标记基因,构建超量表达载体。
16.本发明优选地用于制备球孢白僵菌工程菌株的重组表达载体pbargpe1具有图1所述载体结构。
17.本发明利用随机插入启动子pgpda融合bbeng1基因编码区,获得超量表达bbeng1的工程菌株,获得的工程菌株中bbeng1活性结构域编码区可高水平转录,并且促进了球孢白僵菌生长和分生孢子产生,增强了毒力。可用于制备真菌杀虫剂。
18.本发明优选的另一方法是通过转入组成型启动子pb3(来源于球孢白僵菌3-磷酸甘油醛脱氢酶基因启动子,liao et al.,2008,current microbiology 57:121
–
126;lu et al.,2021,environ microbiol 23(2):1256
–
1274)控制目的基因bbeng1至罗伯茨绿僵菌和蝗绿僵菌野生型菌株,rt-qpcr筛选验证获得超量表达bbeng1工程菌株,所述工程菌株中bbeng1结构域编码区高水平转录。
19.具体的,超量表达bbeng1基因编码区优选的是以真菌基因组成型启动子pb3为实现超量表达的启动子,以除草剂氯磺隆抗性基因sur为标记基因,构建超量表达载体。
20.根据发明的另一方面,制备超量表达bbeng1的罗伯茨绿僵菌和蝗绿僵菌菌株的方法,包括下述步骤:
21.1)以野生型基因组dna为模板扩增bbeng1基因编码区,将扩增片段克隆到骨架载体pk2-pc-sur-tc-pb3(图2),获得超量表达载体;
22.2)将步骤1)获得的表达载体以sur基因为筛选标记,利用农杆菌介导的真菌遗传遗传转化方法(ma et al.,2009,appl microbiol biotechnol 82:891
–
898)分别转入罗伯茨绿僵菌和蝗绿僵菌野生型菌株,用氯磺隆除草剂抗性进行两次筛选获得转化子,验证后的转化子接种于1/4sdy液体培养基中,26℃、200rpm摇床培养3d,提取菌丝rna并反转录为cdna进行qrt-pcr分析筛选超量表达转化子。所获罗伯茨绿僵菌和蝗绿僵菌工程菌株具有促进生长和分生孢子产生和增强毒力特征。
23.本发明优选的另一方法是通过转入组成型启动子pb3控制目的基因mreng1至罗伯
茨绿僵菌野生型菌株,rt-qpcr筛选验证获得超量表达bbeng1工程菌株,所述工程菌株中mreng1结构域编码区高水平转录。
24.具体的,超量表达mreng1基因编码的gh16结构域优选的是以真菌基因组成型启动子pb3为实现超量表达的启动子,以除草剂氯磺隆抗性基因sur为标记基因,构建超量表达载体。
25.根据发明的另一方面,制备超量表达mreng1的罗伯茨绿僵菌菌株的方法,包括下述步骤:
26.1)以罗伯茨绿僵菌野生型基因组dna为模板扩增mreng1基因编码区,将扩增片段克隆到骨架载体pk2-pc-sur-tc-pb3(图2),获得超量表达载体;
27.2)将步骤1)获得的表达载体以sur基因为筛选标记,利用农杆菌介导的真菌遗传遗传转化方法(ma et al.,2009,appl microbiol biotechnol 82:891
–
898)转入罗伯茨绿僵菌野生型菌株,用氯磺隆除草剂抗性进行两次筛选获得转化子,验证后的转化子接种于1/4sdy液体培养基中,26℃、200rpm摇床培养3d,提取菌丝rna并反转录为cdna进行qrt-pcr分析筛选超量表达转化子。所获罗伯茨绿僵菌工程菌株具有促进生长和分生孢子产生和增强毒力特征。
28.本发明优选的另一方法是通过转入组成型启动子pb3控制目的基因mreng1至罗伯茨绿僵菌野生型菌株,rt-qpcr筛选验证获得超量表达mreng1工程菌株,所述工程菌株中mreng1结构域编码区高水平转录。
29.具体的,超量表达mreng1基因编码的gh16结构域优选的是以真菌基因组成型启动子pb3为实现超量表达的启动子,以除草剂氯磺隆抗性基因sur为标记基因,构建超量表达载体。
30.根据发明的另一方面,制备超量表达maeng1的蝗绿僵菌菌株的方法,包括下述步骤:
31.1)以蝗绿僵菌野生型基因组dna为模板扩增maeng1基因编码区,将扩增片段克隆到骨架载体pk2-pc-sur-tc-pb3(图2),获得超量表达载体;
32.2)将步骤1)获得的表达载体以sur基因为筛选标记,利用农杆菌介导的真菌遗传遗传转化方法(ma et al,2009,appl microbiol biotechnol 82:891
–
898)转入蝗绿僵菌野生型菌株,用氯磺隆除草剂抗性进行两次筛选获得转化子,验证后的转化子接种于1/4sdy液体培养基中,26℃、200rpm摇床培养3d,提取菌丝rna并反转录为cdna进行qrt-pcr分析筛选超量表达转化子。所获蝗绿僵菌工程菌株具有促进生长和分生孢子产生和增强毒力特征。
33.本发明优选地用于制备罗伯茨绿僵菌和蝗绿僵菌的表达载体pk2-pc-sur-tc-pb3具有图2所述载体结构。
34.实验结果发现,超量表达bbeng1的球孢白僵菌、罗伯茨绿僵菌和蝗绿僵菌、超量表达mreng1的罗伯茨绿僵菌和超量表达maeng1的蝗绿僵菌菌株在基础和营养丰富的培养基上的生长速率明显快于相应的亲本菌株,而且分生孢子产量显著提高。生物测定结果显示,上述工程菌株毒力显著高于相应的亲本菌株。
35.根据本发明的另一方面,提供一种昆虫生防真菌工程菌株,所述工程菌株具有超量表达的分泌性葡聚糖酶基因eng1。
36.所述昆虫生防真菌选自球孢白僵菌,罗伯茨绿僵菌和蝗绿僵菌;所述昆虫生防真菌分泌性葡聚糖酶基因eng1选自下述一种:来源于球孢白僵菌的分泌性葡聚糖酶基因bbeng1,来源于罗伯茨绿僵菌的bbeng1同源基因mreng1和来源于蝗绿僵菌的bbeng1同源基因maeng1。
37.根据本发明的又一方面,提供一种真菌杀虫剂,包含本发明所述的昆虫生防真菌工程菌株。
38.所述杀虫剂的剂型选自粉剂、乳剂、油剂、微胶囊、混合剂和干菌丝,其中粉剂优选为原粉剂和可湿性粉剂。
39.根据本发明的又一方面,提供昆虫生防真菌分泌性葡聚糖酶基因eng1在制备真菌杀虫剂中的用途,所述昆虫生防真菌分泌性葡聚糖酶基因eng1选自下述一种:来源于球孢白僵菌的分泌性葡聚糖酶基因bbeng1、来源于罗伯茨绿僵菌的bbeng1同源基因mreng1和来源于蝗绿僵菌的bbeng1同源基因maeng1。
40.有益效果:本发明的球孢白僵菌、罗伯茨绿僵菌和蝗绿僵菌工程菌株生长速率明显提升,分生孢子产量增加,毒力显著提升。工程菌株显著提升了生产性状和生防效率(毒力)。
附图说明
41.图1.超量表达骨架载体pbargpe1载体图谱。
42.其中,gpda为来源于构巢曲霉(aspergillus nidulans)3-磷酸甘油醛脱氢酶基因的启动子,为真菌组成型启动子;ptrpc和ttrpc为来源于构巢曲霉色氨酸合成酶基因启动子和终止子;bar为除草剂草丁膦n-乙酰转移酶基因,为真菌转化筛选标记,置于ptrpc控制之下;ampr为氨苄霉素抗性基因,用于大肠杆菌转化筛选标记。
43.图2.超量表达骨架载体pk2-pc-sur-tc-pb3图谱。
44.其中,pc和tc为来源于构巢曲霉色氨酸合成酶基因启动子和终止子;sur为除草剂氯磺隆抗性基因;pb3为来源于球孢白僵菌3-磷酸甘油醛脱氢酶基因的启动子(lu et al.,2021,environ microbiol 23(2):1256
–
1274);kan为卡那霉素抗性基因,用于大肠杆菌转化筛选标记。
45.图3.bbeng1蛋白结构示意图
46.其中,红色区域为信号肽,蓝色区域为gh16_fungal_lam16a_glucanase(gh16)结构域。
△
、
▲
分别表示催化位点和n-糖基化位点。
47.图4.pbargpe1-gfp载体图谱
48.其中,gpda为构巢曲霉三磷酸甘油醛脱氢酶基因gpda的启动子,为真菌组成型启动子;gfp为绿色荧光蛋白编码基因,其置于gpda控制之下;trpc promoter为构巢曲霉色氨酸合成酶启动子,为真菌组成型启动子;bar为除草剂草丁膦n-乙酰转移酶基因,为真菌转化筛选标记,置于trpc promoter控制之下;amp为氨苄青霉素抗性基因,用于大肠杆菌转化筛选标记。
49.图5.bbeng1::gfp融合表达载体图谱
50.该载体编码bbeng1融合gfp蛋白,即将bbeng1基因(4168bp,包括2194bp启动子序列且去除终止密码子)克隆到pbargpe1-gfp载体的ndei和ecorv酶切位点。
51.图6.bbeng1基因表达模式结果
52.a是bbeng1在球孢白僵菌不同形态细胞中的转录水平;b是bbeng1::gfp菌株在不同形态细胞中的gfp荧光观察。ahy、co、lhy、bl和hb分别表示气生菌丝、分生孢子、液体菌丝、芽生孢子和虫菌体;c是rt-qpcr分析bbeng1在不同碳源营养条件下的转录模式。不同碳源条件包括基础培养基(czb)、营养丰富培养基(1/4sdb)、以葡萄糖(glucose)、海藻糖(trehalose)、几丁质(chitin)、葡聚糖(dextran)置换察氏培养基中的蔗糖的培养基、0.167mg/ml昆虫体壁(cuticle)和0.5ml/l昆虫血淋巴(heamolymph),诱导时间均为6h;d是bbeng1::egfp在不同碳源培养基诱导培养的gfp荧光观察;e是rt-qpcr分析昆虫血淋巴和昆虫体壁诱导培养条件下bbeng1的时间进程转录水平;f是在昆虫营养物质(血淋巴和体壁)诱导培养不同时间bbeng1::gfp菌株的gfp荧光观察。bar=5μm。
53.图7.bbeng1在球孢白僵菌的细胞分布及分泌特征
54.a是bbeng1::gfp在球孢白僵菌虫菌体形态不同时间的gfp荧光观察;b是bbeng1::gfp菌丝体表面的gfp荧光分布在fm4-64染色的细胞膜外;c是western blotting检测球孢白僵菌野生型菌株(wt)在昆虫血淋巴和体外昆虫营养物质(角质层或血淋巴)中诱导培养的上清液中分泌的bbeng1,一抗为bbeng1多克隆兔抗,二抗为羊抗兔;bar=10μm。
55.图8.bbeng1基因敲除、回复互补及超量表达策略及分子验证
56.a是bbeng1基因敲除载体构建策略图。标记“x”的交叉事件表示同源重组,bbeng1的特定区域被bar基因替换;b是超量表达载体和回复互补载体构建策略。以pbargpe1为骨架载体,将bbeng1的编码框置于组成型启动子gpda下游构建超量表达载体。c是回复互补载体pcb-bbeng1以sur为筛选标记;d是以s1/s2为筛选引物pcr扩增筛选验证敲除及回复菌株;e是rt-pcr验证敲除及回复菌株;f是real time rt-pcr检测wt及bbeng1超量表达转化子的bbeng1的转录水平;g是δbbeng1菌株和comp菌株的southern blot检测。基因组使用hind iii消化,δbbeng1使用bar探针,comp使用sur探针。
57.图9.pk2-gus载体图谱
58.其中,ptrpc和ttrpc为来源于构巢曲霉色氨酸合成酶基因启动子和终止子;gus为β-葡萄糖苷酸酶基因,用于随机插入的筛选。
59.图10.pk2-ptrpc-sur-ttrpc载体图谱
60.其中,ptrpc和ttrpc为来源于构巢曲霉色氨酸合成酶基因启动子和终止子;sur为除草剂氯嘧磺隆抗性基因,用于真菌转化筛选标记。
61.图11.毕赤酵母表达bbeng1验证及酶学特性
62.a是纯化bbeng1的sds-page分析;b是western blot检测bbeng1结合多种不溶性多糖;c是使用image软件测量bbeng1结合不溶性多糖的免疫印迹定量结果;d和e为fitc标记的bbeng1蛋白(bbeng1
fitc
)与几种不溶性多糖的结合情况的fitc荧光观察和fitc荧光强度定量结果;f是bbeng1对几种多糖的水解反应tlc图谱。g1~g5为标准糖:葡萄糖(g1)、麦芽糖(g2)、麦芽三糖(g3)、麦芽四糖(g4)、麦芽五糖(g5);g是bbeng1对几种多糖的降解产物的hplc分析。这些试验中使用的bbeng1蛋白浓度为20μg/ml。不溶性多糖包括:大麦β-葡聚糖(barleyβ-glucan)、石耳葡聚糖(pustulan)、羟甲基纤维素钠(cmc-na)、茯苓聚糖(pachyma)、壳聚糖(chitosan)和昆虫体壁(cuticle)(1%,w/v);可溶性多糖包括:酵母葡聚糖(yeast glucan)、海带多糖(laminarin)、右旋糖酐(dextran)(1%,w/v)。
63.图12.hplc验证bbeng1对昆虫营养的水解活性。高效液相色谱(hplc)检测bbeng1处理昆虫体壁、血淋巴和海藻糖的降解产物。
64.图13.球孢白僵菌野生菌株(wt)、bbeng1基因敲除(δbbeng1)、互补(comp)及超量表达(bbeng1
oe
)菌株的菌落生长、产孢及形态和孢子萌发率。
65.a是在不同碳源的培养基上的菌落形态;b是在不同碳源的培养基上的生长速率;c是产孢结构;d是在基础培养基(cza)和富营养培养基(1/4sday)上的分生孢子产量;e是分生孢子形态;f是不同面积的分生孢子丰度统计;g是各菌株分生孢子萌发率曲线图。
66.图14.球孢白僵菌野生菌株(wt)、bbeng1基因敲除(δbbeng1)、互补(comp)及超量表达(bbeng1
oe
)菌株的菌丝形态。
67.a是基础培养基上(cza)菌丝形态;b是萌发菌丝细胞壁专性染料荧光增白剂(cfw)染色的荧光图像;c是双向发芽率;d是荧光增白剂(cfw)和核特异性染料碘化丙啶(pi)染色的萌发菌丝的荧光图像;e是每50μm菌丝的隔膜和细胞核数量统计;bar=20μm。
68.图15.球孢白僵菌野生菌株(wt)、bbeng1基因敲除(δbbeng1)、互补(comp)及超量表达(bbeng1
oe
)菌株细胞壁结构及组成。
69.a是各菌株虫菌体透射电镜(tem)照片;b是真菌细胞壁的厚度;c是利用荧光增白剂calcofluor white stain(cfw)和苯胺蓝aniline blue染色,根据荧光值分别测定各菌株虫菌体细胞壁中几丁质和葡聚糖水平。d是使用image j计算徕卡sp8激光共聚焦显微镜采集荧光图片的荧光强度;bar=5μm。
70.图16.球孢白僵菌野生菌株(wt)、bbeng1基因敲除(δbbeng1)、互补(comp)及超量表达(bbeng1
oe
)菌株生物测定。
71.a为通过体壁接种1ml浓度为3
×
107孢子/ml的分生孢子后试虫的存活率趋势;b为微量注射2μl浓度为5
×
106孢子/ml的孢子悬浮液至试虫血腔后,试虫的存活率趋势;c为体壁接种后大蜡螟形态及菌丝穿出及产孢;d为注射接种僵虫形态及菌丝穿出及产孢。
72.图17.球孢白僵菌野生菌株(wt)、bbeng1基因敲除(δbbeng1)、互补(comp)及超量表达(bbeng1
oe
)菌株接种后昆虫的免疫反应及在虫体内的增殖。
73.a是体壁接种方式接种1ml浓度为3
×
107/ml的分生孢子后试虫体表的黑化反应;b是微量注射接种2μl浓度为5
×
106/ml的分生孢子到试虫血腔,试虫体表黑化反应;c是微量注射接种2μl浓度为5
×
106/ml的各菌株分生孢子后昆虫血腔内真菌生长发育及免疫反应显微观察;bar=20μm;d是微量注射接种36h、48h和60h后虫菌体定量数据。采用qpcr方法进行。
74.图18.球孢白僵菌野生菌株(wt)、bbeng1基因敲除(δbbeng1)、互补(comp)及超量表达(bbeng1
oe
)菌株的体液免疫反应。
75.a是注射接种各菌株后大蜡螟血淋巴样品中的po活性曲线;b是微量注射接种各菌株后大蜡螟血淋巴中h2o2(ros)水平的变化;c是注射接种12h和24h后大蜡螟脂肪体抗菌肽相关基因和toll途径基因的转录模式。以大蜡螟的肌动蛋白actin为内参。接种量均为5
×
106孢子/ml,2μl/虫。
76.图19.球孢白僵菌野生菌株(wt)、bbeng1基因敲除(δbbeng1)、互补(comp)及超量表达(bbeng1
oe
)虫菌体表位及病原相关分子模式
77.a是alexa fluor 488标记的凝集素菌丝体表面碳水化合物表位和β-1,3-葡聚糖
水平的荧光图像。b是使用image软件测量至少100个单个细胞的平均荧光。
78.图20.pk2-pc-sur-tc-pb3::bbeng1载体图谱
79.将bbeng1基因编码区(1767bp)克隆到pk2-pc-sur-tc-pb3的bamhⅰ和ecorv位点,置于组成型启动子pb3控制下,构建超量表达载体。
80.图21.超量表达bbeng1、mreng1和maeng1载体构建示意图及菌株筛选超量表达载体构建策略。
81.a以pk2-pc-sur-tc-pb3为骨架载体,将bbeng1、mreng1和maeng1的编码框置于组成型启动子pb3下游构建超量表达载体。b是real time rt-pcr检测罗伯茨绿僵菌中超量表达bbeng1转化子的bbeng1的转录水平;c是real time rt-pcr检测蝗绿僵菌中超量表达bbeng1转化子的bbeng1的转录水平;d是real time rt-pcr检测罗伯茨绿僵菌wt及超量表达mreng1转化子的mreng1的转录水平;e是real time rt-pcr检测蝗绿僵菌wt及超量表达maeng1转化子的maeng1的转录水平。
82.图22.超量表达bbeng1、mreng1和/或maeng1的罗伯茨绿僵菌和蝗绿僵菌与其野生菌株生长、产孢及孢子萌发
83.a是各菌株在基础培养基(cza)和富营养培养基(pda)培养基上的菌落形态;b是各菌株在液体培养基(1/4sdy)生长状态;c是各菌株在基础培养基(cza)上的分生孢子产量;d是各菌株在基础培养基(cza)上分生孢子萌发率曲线图及萌发中时(gt
50
)。
84.图23.超量表达bbeng1、mreng1和/或maeng1的罗伯茨绿僵菌和蝗绿僵菌与其野生菌株生物测定
85.a为通过体壁接种方式分别接种1ml浓度为3
×
107孢子/ml罗伯茨绿僵菌野生型(wt)、超量表达bbeng1的罗伯茨绿僵菌菌株(mr-bbeng1
oe
)和超量表达mreng1的罗伯茨绿僵菌株(mr-mreng1
oe
)分生孢子后大蜡螟幼虫的存活率趋势;b为微量注射接种罗伯茨绿僵菌各菌株2μl浓度为5
×
106孢子/ml的孢子悬浮液至大蜡螟血腔后,试虫的存活率趋势;c为通过背板点滴方式分别接种5μl浓度为3
×
107孢子/ml的蝗绿僵菌野生型(wt)、超量表达bbeng1蝗绿僵菌(ma-bbeng1
oe
)和超量表达maeng1蝗绿僵菌(ma-maeng1
oe
)分生孢子后东亚飞蝗的存活率趋势;d为微量注射蝗绿僵菌各菌株2μl浓度为5
×
106孢子/ml的孢子悬浮液至大蜡螟血腔后,试虫的存活率趋势。
86.图24.mreng1蛋白结构图
87.图25.mreng1和maeng1在不同形态细胞中的表达模式
88.rt-qpcr分析mreng1在罗伯茨绿僵菌不同形态细胞中的表达模式,以罗伯茨绿僵菌三磷酸甘油醛脱氢酶基因mrgpd为参比基因;maeng1在蝗绿僵菌不同形态细胞中的表达模式,以蝗绿僵菌三磷酸甘油醛脱氢酶基因magpd为参比基因。
89.图26.pk2-pc-sur-tc-pb3::mreng1载体图谱
90.将mreng1基因编码区(1487bp)克隆到pk2-pc-sur-tc-pb3的bamhⅰ和ecorv位点,置于组成型启动子pb3控制下,构建超量表达载体。
91.图27.maeng1蛋白结构
92.图28.pk2-pc-sur-tc-pb3::maeng1载体图谱
93.将maeng1基因编码区(1520bp)克隆到pk2-pc-sur-tc-pb3的bamhⅰ和ecorv位点,置于组成型启动子pb3控制下,构建超量表达载体。
94.图29.bbeng1与mreng1、maeng1的共同点
95.其中,a是bbeng1、mreng1和maeng1结构的示意图,sp表示信号肽,gh16表示gh16_fungal_lam16a_glucanase结构域,
▲
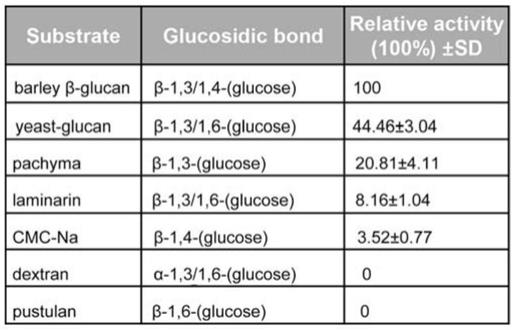
、
△
分别表示催化位点和活性位点;b是bbeng1、mreng1和maeng1结构域区域氨基酸序列比对结果(使用clustal w方法),sp表示信号肽,gh16表示gh16_fungal_lam16a_glucanase结构域,*表示半胱氨酸残基;c是bbeng1在球孢白僵菌不同形态细胞中的转录水平,参比基因为18s rrna(gen-bank id:eu334679);d是以mrgpd(genbank id:19261961)为参比基因检测mreng1在罗伯茨绿僵菌不同形态细胞中的转录水平;e是以magpd(gen-bank id:19253895)为参比基因检测maeng1在蝗绿僵菌不同形态细胞中的转录水平;ahy、co、lhy、bl和hb分别表示气生菌丝、分生孢子、液体菌丝、芽生孢子和虫菌体。
具体实施方式
96.通过以下实施例可以进一步理解本发明的优点和特点,不应该理解为是对本发明范围的限制。
97.以下实施例中所用仪器和试剂,除特殊说明以外均为普通市售。
98.超量表达的目的基因描述(昆虫生防真菌分泌性葡聚糖酶基因eng1):
99.bbeng1为分离于球孢白僵菌侵入昆虫体内繁殖菌体(虫菌体)特异的表面蛋白库,在基因组注释为concanavalin a-like lectin/glucanase(bba_04753)(yang et al.,2015,fungal genet biol 99:13-25)。bbeng1编码区(1974bp)含有3个内含子,编码一个含有588个氨基酸残基(64.4kda)的蛋白质。
100.以球孢白僵菌bbeng1氨基酸序列为探针,利用blastp搜索罗伯茨绿僵菌基因组(genbank:gca_000187425.2)和蝗绿僵菌基因组(genbank:gca_000187405.1),分别克隆获得bbeng1同源蛋白编码基因mreng1(maa_09026,相似性为52.9%)和maeng1(mac_06610,相似性55.6%),基因组分别注释为concanavalin a-like lectin/glucanase和β-1,3-endoglucanase基因,编码蛋白分别有432(48.1kda)和448个氨基酸残基(49.9kda)组成。
101.采用uniprot网站(https://www.uniprot.org/)中的blastp程序进行蛋白结构域分析显示,bbeng1、mreng1和maeng1具有相似的结构,包含一个保守的gh16(gh16_fungal_lam16a_glucanase)结构域),n端包含一个典型的信号肽序列,无跨膜结构,无gpi锚定位点,含有多个糖基化位点,包含多个半胱氨酸残基及是受体结合和(或)催化位点(图29)。表达分析显示,bbeng1、mreng1和mreng1三个基因具有相同的表达模式,均在昆虫体内增殖的虫菌体特异表达。
102.【实施例1】
103.1.bbeng1蛋白的结构域分析
104.bbeng1分离于昆虫球孢白僵菌侵入昆虫体内繁殖菌体(虫菌体)特异的表面蛋白库,在基因组注释为concanavalin a-like lectin/glucanase(bba_04753)(yang et al.,2015,fungal genet biol 99:13-25)。bbeng1编码区(1974bp)含有3个内含子,编码一个含有588个氨基酸残基(64.4kda)的蛋白质。采用uniprot网站(https://www.uniprot.org/)中的blastp程序进行蛋白结构域分析,bbeng1包含一个gh16(gh16_fungal_lam16a_glucanase)结构域),位于氨基端第52至293氨基酸。n端包括一个信号肽序列,无跨膜结构,
无gpi锚定位点,含有多个糖基化位点,包含多个半胱氨酸残基,推测具有二硫键。五个假定的活性位点分别位于第126-130氨基酸、第141-146氨基酸、第155-157氨基酸、第203氨基酸和第277-279氨基酸,是受体结合和(或)催化位点(图3)。
105.2.荧光标签菌株bbeng1::gfp的获取
106.载体构建:以球孢白僵菌基因组dna为模板,利用引物对lf/lr扩增bbeng1基因(4168bp,包括2194bp启动子序列且去除终止密码子),将其克隆到pbargpe1-gfp载体(图4)ndei和ecorv酶切位点,置换组成型启动子pgpda,构建成bbeng1::gfp融合基因(seq id no.1)表达载体(图5)。
107.bbeng1基因扩增体系如下:2
×
phanta max buffer 12.5μl,dntp mix 0.5μl,phanata max super-fidelity dna polymerase 0.5μl,5μmol/l引物lf和lr各1μl,球孢白僵菌野生型基因组20ng,用水补足至25μl体系。扩增程序为:95℃5min;95℃30s,55℃30s,72℃4min,35个循环;72℃延伸10min。
108.遗传转化及转化子验证:用peg介导的芽孢子转化法(ying et al.,2006,appl microbiol biotechnol 72:206
–
210)将荧光标签载体bbeng1::gfp转入球孢白僵菌野生菌株。在含200μg/ml除草剂草甘膦(glufosinate)和500μg/ml的头孢克圬的察氏培养基(czapek-dox agar)平板上筛选抗性菌落。裂解液裂解转化子菌丝,取3μl裂解液作为模板,以bbeng1::gfp质粒为阳性对照,采用引物rt1和gfp-r进行pcr扩增验证。
109.lf1:5'-cggtatttcacaccgcatatgtagctgatgctctccgcgtc-3'(seq id no.2)
110.lr1:5'-gcccttgctcaccatgatatcggcacggcagatttggttgg-3'(seq id no.3)
111.rt1:5'-ctacaagccagagtcgtcctc-3'(seq id no.4)
112.gfp-r:5'-tctcgttggggtctttgctc-3'(seq id no.5)
113.3.bbeng1表达特性
114.为探究bbeng1的表达模式,采用rt-qpcr及荧光标签技术(bbeng1::gfp)分析bbeng1的表达特性。不同形态细胞的收集、不同碳源培养基诱导条件和昆虫营养诱导条件的菌丝样品收集方式如下:
115.气生菌丝及分生孢子收集:用0.05%(v/v)的tween-80配制浓度为1
×
107孢子/ml的球孢白僵菌分生孢子孢悬液,涂布法接种100μl至铺有玻璃纸的1/4sday固体培养基,26℃倒置培养3d收集气生菌丝。培养至10d,收集玻璃纸上菌体,0.05%(v/v)tween-80充分悬浮,用4层擦镜纸过滤除去菌丝,滤液离心,用无菌ddh2o洗涤2次,即为分生孢子样品。
116.液生菌丝及芽生孢子收集:配制浓度为1
×
107孢子/ml的球孢白僵菌分生孢子孢悬液,接种100μl至50ml 1/4sdy液体培养基,26℃、200rpm培养2d,去除培养液,固体沉淀即液生菌丝。相同操作,继续培养至4d,用4层擦镜纸过滤除去菌丝,滤液离心,用无菌ddh2o洗涤2次,即为芽生孢子样品。
117.虫菌体收集:参照yang et al.(fungal genet biol,2017,99:13
–
25)方法进行,配制浓度为1
×
107孢子/ml的分生孢子孢悬液,微量注射2μl至大蜡螟三龄幼虫,冰上收集血腔增殖2d的虫菌体,使用预冷的0.1m pbs洗涤3次,10000rpm离心5min。预冷的0.1m pbs(1mm cacl2,ph 8.0)重悬备用。取10ml离心管,底部缓慢加入3ml 50%centricoll,待其液面平整后,缓慢加入3ml 25%centricoll分离液,最后在分离液上轻轻加入1ml细胞悬浮液,4℃、10000rpm离心5min,收集沉淀即为虫菌体。
118.不同碳源诱导培养的样品收集:接种100μl浓度1
×
107孢子/ml的球孢白僵菌bbeng1::gfp分生孢子孢悬液至200ml的1/4sdy培养基,培养3.5d,用4层擦镜纸过滤除去菌丝,滤液离心,用无菌ddh2o洗涤2次,平均分6份置于30ml各诱导培养基,诱导培养6h,收集诱导菌体。同时各取50μl用于显微观察。诱导培养基包括基础培养基(czb)、营养丰富培养基(1/4sdb)、以葡萄糖(glucose)、海藻糖(trehalose)、几丁质(chitin)、葡聚糖(dextran)置换察氏培养基中的蔗糖的培养基、包含0.167mg/ml昆虫体壁(cuticle)和0.5ml/l昆虫血淋巴(heamolymph)的基础盐培养基(bs)。
119.昆虫营养诱导不同时间样品收集:接种50μl浓度1
×
107孢子/ml的球孢白僵菌bbeng1::gfp分生孢子孢悬液至30ml 1/4sdy培养基,培养3.5d,用4层擦镜纸过滤除去菌丝,滤液离心,用无菌ddh2o洗涤2次,分为2份,分别置于30ml包含0.167mg/ml昆虫体壁(cuticle)和0.5ml/l昆虫血淋巴(heamolymph)的基础盐培养基,收集0、4、8和12h的菌体用于提取rna,同时取样显微观察。
120.rna提取按照easyspin植物rna快速提取试剂盒(北京艾德生物科技有限公司)方法进行。紫外分光光度计定量rna。取1μg rna利用oligo(dt)引物反转录成cdna第一链,反转录参照试剂盒(oligo(dt)-primed cdna synthesis kit(mbi fermentas)说明书。将合成的cdna第一链稀释成10ng/μl,以18s rrna(gen-bank id:eu334679)为参比基因,rt-qpcr检测bbeng1的转录。扩增体系如下:2
×
sybr buffer 5μl,5pmol/l引物各1μl,稀释的cdna链模板3μl。扩增程序如下:95℃2min;95℃5s,60℃30s,39个循环;65℃-95℃升高0.5℃/5s;扩增18s rrna和bbeng1转录本的引物对分别为18s rrna-f/18s rrna-r和rt1/rt2。
121.转录水平检测显示,bbeng1仅在虫菌体形态下高水平转录,而在腐生形态的细胞水平转录极低或者不转录(图6a)。利用荧光标签菌株bbeng1::gfp观察gfp荧光,检测bbeng1的转录及蛋白表达水平,也验证了bbeng1蛋白仅在虫菌体形态下表达(图6b)。
122.为明确bbeng1与碳源利用的关系,利用rt-qpcr检测bbeng1在不同碳源条件下的表达模式。结果显示,在基础培养基(czb)、以不同碳源置换察氏培养基(czb)中的蔗糖的培养基、营养丰富培养基(1/4sdb)培养条件下诱导6h,bbeng1均低水平转录,而在以昆虫体壁、昆虫血淋巴为唯一营养的培养基中诱导6h则高水平转录(图6c),bbeng1::gfp菌株的gfp荧光观察结果与rt-qpcr结果一致,且gfp荧光主要分布在新生长的菌体部位(图6d)。分别以昆虫体壁和昆虫血淋巴为唯一营养培养不同时间检测发现,bbeng1在昆虫营养诱导4h开始转录,且随诱导时间延时,转录水平显著上升(图6e)。而且,bbeng1::gfp菌株的gfp荧光也主要分布在新生长的菌体部位(图6f)。由此推测,bbeng1与昆虫营养利用相关。
123.18s rrna-f:5'-acgggtaacggagggttagg-3'(seq id no.6)
124.18s rrna-r:5'-agtacacgcggtgaggcgga-3'(seq id no.7)
125.rt2:5'-aggtgccctgctggat-3'(seq id no.8)
126.3.bbeng1在球孢白僵菌的细胞分布及分泌特征
127.为探究bbeng1在细胞的分布及分泌特性,观察bbeng1::gfp的虫菌体形态gfp荧光分布,利用荧光膜染料fm4-64(thermo)染色细胞膜,发现bbeng1分布在细胞膜外—细胞壁和/或细胞表面(图7a和b)。分别收集菌体在昆虫血腔增殖时的血淋巴和体外昆虫诱导培养菌体的上清液,滤去菌体,沉淀蛋白,以bbeng1多克隆兔抗为一抗和羊抗兔的二抗进行western blotting检测。结果显示,在昆虫血淋巴和体外昆虫营养诱导培养液中均检测到
synthesis kit(mbi fermentas)说明书。将合成的cdna第一链稀释成10ng/μl,以18s rrna(gen-bank id:eu334679)为参比基因利用rt-pcr扩增bbeng1编码区的转录。扩增18s rrna和bbeng1引物分别为18s rrna-f/18s rrna-r和rt1/rt2。检测扩增体系如下:2
×
phanta max buffer 12.5μl,dntp mix 0.5μl,phanata max super-fidelity dna polymerase 0.5μl,5μmol/l引物各1μl,稀释的cdna一链模板1μl,用水补足至25μl体系。扩增程序如下:95℃5min;95℃30s,55℃30s,72℃30s,25个循环;72℃延伸10min。扩增产物在1.0%(w/v)的琼脂糖凝胶电泳检测bbeng1的转录情况。扩增18s rrna和bbeng1的引物对分别为18s rrna-f/18s rrna-r和rt1/rt1。结果表明,bbeng1在敲除突变株无法正常转录(图8e)。
135.提取基因破坏突变体基因组dna,利用hindiii酶切后于1.0%的琼脂糖凝胶电泳分离,通过高盐转膜法转移到尼龙膜(hybondtm-n nylon membrane,amersham biosciences,usa),然后用引物b3/b4扩增bar基因片(411bp)(seq id no.11)为探针,用地高辛标记探针进行杂交,杂交为单拷贝插入的转化子为进一步确认的单拷贝的基因破坏转化子(图8g)。具体操作参照地高辛标记试剂盒(dig-high prime dna labeling and detection starter kit i,roche)。
136.l1:5'-acatgattacgaattcgggtgtctgttttgtgtgcg-3'(seq id no.12)
137.l2:5'-caatgtcatcttctgtcgacctgtatggcgtgtgaggcaa-3'(seq id no.13)
138.r1:5'-tgcccgtcaccgagatctaagtacaattccggatcggcca-3'(seq id no.14)
139.r2:5'-caacactagtggatccggttctcggcaacgtactga-3'(seq id no.15)
140.b1:5'-ttgcctcacacgccatacaggtcgacagaagatgacattg-3'(seq id no.16)
141.b2:5'-tggccgatccggaattgtacttagatctcggtgacgggca-3'(seq id no.17)
142.b3:5'-accttcttaagttcgccctt-3'(seq id no.18)
143.b4:5'-gtagagcgtggagcccagt-3'(seq id no.19)
144.s3:5'-tggtagcactctcgcagttg-3'(seq id no.20)
145.s4:5'-ctcaaagtccacgcccagat-3'(seq id no.21)
146.2.回复互补bbeng1破坏突变体
147.以球孢白僵菌基因组dna为模板,利用引物prc1/prc2(分别在prc1和prc2的5'-端引入bamhi和xbai酶切位点,引物序列附后)扩增bbeng1基因(包括启动子序列、编码区和终止子序列,共4118bp)。用bamhi和xbai酶切扩增片段,克隆到用相同酶切的载体pk2-ptrpc-sur-ttrpc载体(图10)上,形成载体pk2-sur-bbeng1(图8c),该载体携带除草剂氯嘧磺隆(chlorimuronethyl)抗性基因sur。将表达载体pk2-sur-bbeng1转入根癌农杆菌agl-1菌株,转化参照fang et al.方法(2004,j invertebr pathol 85:18-24)。然后利用根癌农杆菌介导法转化至球孢白僵菌δbbeng1突变体分生孢子(ma et al.,2009,appl microbiol biotechnol 82:891
–
898),转化后在含有4μg/ml chlorimuronethyl的察氏培养基(czapek-dox agar)上筛选抗性菌落。提取抗性菌落dna,利用引物s3/s4验证转化子。若将bbeng1成功导入δbbeng1突变体,则扩增出两条带,一条为含有bar基因的部分同源重组元件(1263bp),另一条为野生型基因部分片段(812bp)(图8d)。
148.用rt-pcr验证转化子中bbeng1是否回复正常转录。rt-pcr操作如下:用0.05%(vol/vol)tween-80配制浓度为1
×
107孢子/ml分生孢子悬浮液,微量注射接种于大蜡螟三龄幼虫,2μl/虫,26℃培养48h,收集虫菌体提取rna,反转录后合成cdna第一链。以野生菌株
(wt)相同处理为对照进行rt-pcr表达分析。rna提取按照easyspin植物rna快速提取试剂盒(北京艾德生物科技有限公司)方法进行。紫外分光光度计定量rna。取2μg rna利用oligo(dt)引物反转录成cdna第一链,反转录参照试剂盒(oligo(dt)-primed cdna synthesis kit(mbi fermentas)说明书。将合成的cdna第一链稀释成10ng/μl,以18s rrna(gen-bank id:eu334679)为参比基因,利用rt-pcr扩增bbeng1编码区的转录。扩增18s rrna和bbeng1引物分别为18s rrna-f/18s rrna-r和rt1/rt2。检测扩增体系如下:2
×
phanta max buffer 12.5μl,dntp mix 0.5μl,phanata max super-fidelity dna polymerase 0.5μl,5μmol/l引物各1μl,稀释的cdna一链模板1μl,用水补足至25μl体系。扩增程序如下:95℃5min;95℃30s,55℃30s,72℃30s,25个循环;72℃延伸10min。扩增产物在1.0%(w/v)的琼脂糖凝胶电泳检测bbeng1的转录情况。结果表明,bbeng1在回复互补转化子中能够正常转录,转录水平与野生型一致(图8)。
149.提取回复互补转化子基因组dna,利用hindiii酶切后于1.0%的琼脂糖凝胶电泳分离,通过高盐转膜法转移到尼龙膜(hybond
tm-n nylon membrane,amersham biosciences,usa),然后利用引物sur1/sur2扩增sur片段(380bp)(seq id no.22)为探针,用地高辛标记进行杂交,杂交结果为单拷贝插入的转化子(图8g)。具体操作参照地高辛标记试剂盒(dig-high prime dna labeling and detection starter kit i,roche)。
150.prc1:5'-tgctctcacgtcgacggatccattggcagaggtgtctccac-3'(seq id no.23)
151.prc2:5'-tgcctgcaggtcgactctagattaggcacggcagatttggt-3'(seq id no.24)
152.sur1:5'-agtgtgctgaggagggctat-3'(seq id no.25)
153.sur2:5'-acacggtcatcgaagcggcca-3'(seq id no.26)
154.5.超量表达bbeng1球孢白僵菌菌株构建
155.利用组成型启动子增加球孢白僵菌bbeng1的表达。
156.构建超量表达bbeng1的球孢白僵菌菌株载体策略如下:利用构巢曲霉组成型启动子gpda融合目的基因编码区序列,经遗传转化导入球孢白僵菌,组成型启动子gpda增加bbeng1的表达,利用rt-qpcr扩增筛选获得bbeng1超量表达转化子。
157.具体操作如下:
158.以球孢白僵菌基因组dna为模板,利用引物对oe-f1/oe-r1扩增bbeng1基因编码区(1974bp)(seq id no.29),将其克隆到pbargpe1(图1)的bamhⅰ和ecorⅰ位点,置于组成型启动子gpda控制下,构建超量表达载体,以bar基因为筛选标记。利用peg4000介导球孢白僵菌芽生孢子的遗传转化方法(ying et al.,2006,appl microbiol biotechnol 72:206
–
210),用草胺膦除草剂(200μg/ml)抗性进行两次筛选获得转化子,验证后的转化子接种于1/4sdy液体培养基中,26℃、200rpm摇床培养3d,提取菌丝rna并反转录为cdna进行rt-qpcr分析筛选超量表达转化子。目的片段扩增体系如下:2
×
phanta max buffer 12.5μl,dntp mix 0.5μl,phanata max super-fidelity dna polymerase 0.5μl,5μmol/l引物oe-f1/oe-r1各1μl,球孢白僵菌野生型基因组20ng,用水补足至25μl体系。扩增程序为:95℃5min;95℃30s,55℃30s,72℃2min,35个循环;72℃延伸10min。扩增产物在1.0%(w/v)的琼脂糖凝胶电泳,回收扩增片段测序验证。然后用bamhi和ecori酶切融合片段,连接于用相同酶切后的pbargpe1,形成超量表达载体pbargpe1::bbeng1(图8)。
159.采用peg4000介导芽生孢子的遗传转化方法转化将表达载体pbargpe1::bbeng1转
入球孢白僵菌野生型菌株,在含200μg/ml除草剂草甘膦(glufosinate)的察氏培养基(czapek-dox agar)平板上筛选抗性菌落。提取抗性转化子dna,利用引物s1/s2扩增筛选基因超量突变体,若插入转化子则扩增出部分gpda和部分目的条带(652bp),未成功转入则无法扩增出条带,根据该方案,筛选到多个转化成功的转化子。
160.接种野生菌株(wt)和超量转化子至1/4sdy液体培养基,26℃培养60h,收集菌丝并提取rna。rna提取按照easyspin植物rna快速提取试剂盒(北京艾德生物科技有限公司)方法进行。紫外分光光度计定量rna。取1μg rna利用oligo(dt)引物反转录成cdna第一链,反转录参照试剂盒(oligo(dt)-primed cdna synthesis kit(mbi fermentas)说明书。将合成的cdna第一链稀释成10ng/μl,以18s rrna(gen-bank id:eu334679)为参比基因,rt-qpcr检测bbeng1的转录。扩增体系如下:2
×
sybr buffer 5μl,5pmol/l引物各1μl,稀释的cdna链模板3μl。扩增程序如下:95℃2min;95℃5s,60℃30s,39个循环;65℃-95℃升高0.5℃/5s;扩增18s rrna和bbeng1转录本的引物对分别为18s rrna-f/18s rrna-r和rt1/rt2。结果表明,筛选到转化子bbeng1转录水平提高了2.17-235.55倍(图8f)
161.oe-f1:5'-cgcggatccatgccgtcactcatttcgtg-3'(seq id no.27)
162.oe-r1:5'-ggaattcggcacggcagatttggttgg-3'(seq id no.28)
163.s1:5'-tgagaaggttttgggacgct-3'(seq id no.30)
164.s2:5'-cgaccatttggattggacgc-3'(seq id no.31)
165.6.酵母表达bbeng1与蛋白纯化
166.酵母表达载体构建:以球孢白僵菌野生型虫菌体cdna为模板,利用com1和com2引物扩增去除信号肽序列的bbeng1的编码区cdna(1707bp)(seq id no.34),分别在cdna的5'端和3'端引入ecori和noti酶切位点,并将6个串联的his标签通过引物分别引入cdna序列的两端,胶回收获得pcr产物。利用ecori和noti酶切位点将扩增产物克隆到ppic9k载体(invitrogen,carlsbad,ca,usa)相同酶切位点。测序验证后用于酵母遗传转化。
167.毕赤酵母转化:参照毕赤酵母pichia expression kit(invitrogen)说明书,利用电转化法将质粒导入甲醇诱导型毕赤酵母(pichia pastoris)gs115菌株(invitrogen),于选择培养基md培养基(含1.34%(w/v)ynb(yeast nitrogen base)(gifco,ks,usa)、40mg/ml biotin(invitrogen)、2%(w/v)glucose和1.5%(w/v)agar)上培养。将生长的重组菌株在ypd液体培养基(含1%(w/v)yeast extract、2%(w/v)peptone、2%(w/v)glucose)上连续继代培养两次。然后将转化子转接到含有1.5mg/ml geneticin(g418)(takara,dalian,china)的ypd培养基上筛选多拷贝转化子。筛选的转化子进一步利用引物com1和com2扩增验证,获得阳性转化子。引物序列附后。com15'-ccggaattcatgcatcatcaccatcaccataagtattcgctgtcccaaa-3'(seq id no.32)com25'-aaggaaaaaagcggccgcttaatggtgatggtgatgatgggcacggcagatttggttg-3'(seq id no.33)
168.蛋白诱导表达及纯化:接种筛选的重组酵母菌株于25ml bmgy培养基(含2%(w/v)peptone,1%(w/v)yeast extract,100nmol/l potassium phosphate(ph 6.0),biotin 40mg/ml和1%(v/v)glycerol),在28℃、180rpm条件下摇瓶培养48h(od
600
≈2.0),放大接种于500ml用0.5%(v/v)甲醇(methanol)置换1%(v/v)甘油(glycerol)的bmgy培养诱导目标蛋白表达。诱导培养6天后,6000g离心去除菌体后收集上清液,然后用0.45mm的滤膜除去酵母细胞。用80%(nh4)2so4于4℃过夜沉淀蛋白质,于12000g、4℃离心30min收集沉淀的蛋白
质。用5ml 0.2m pbs(ph 7.5)溶解沉淀蛋白质,然后于脱盐柱hipreptm 26/10desalting column(ge healthcare life sciences)脱盐。脱盐后的样品用magene histm protein purification system(promega)纯化目标蛋白。以牛血清白蛋白(bsa)为标准曲线,用bradford法对纯化的bbeng1浓度进行标准化。纯化后的蛋白使用十二烷基硫酸钠聚丙烯酰胺凝胶电泳(sds-page)电泳和western blotting进行纯化蛋白确定。电泳结果显示,获得正确的bbeng1(64.4kda)蛋白(图11)。
169.7.bbeng1酶学功能
170.利用纯化的bbeng1测试了其与多种不溶性多糖的结合特性(cen et al.,2017,plos pathog 13(9):e1006604)。蛋白与不同底物共孵育后,通过sds-page和western blotting电泳检测未结合蛋白(上清液)和结合蛋白(沉淀于多糖)。结果表明,bbeng1对大麦葡聚糖(barleyβ-glucan)、纤维素钠(cmc-na)、昆虫体壁(cuticle)结合能力强,对石耳葡聚糖(pustulan)和茯苓聚糖(pachyma)有较弱结合能力,但不与甲壳素(chitosan)结合(图11)。采用异硫氰酸(fitc)标记bbeng1蛋白bbeng1
fitc
与不溶性多糖及昆虫体壁颗粒的结合试验结果与凝胶印记结果一致(图11)。
171.β-1,3/4-葡聚糖酶水解活性测定采用二硝基水杨酸(dns)法进行(miller,anal chem,1959,31:426-426)。具体如下:
172.dns法测定还原糖含量方法:90μl含有10mm多糖底物的50mm乙酸钠缓冲液(ph 6.0)中加入10μl蛋白酶液(0.02m pbs溶解,2μg),在37℃孵育1h后,加入200μl dns缓冲液,沸水浴5min,冰上3min后,检测540nm处吸光度。标准曲线制备:使用50mm乙酸钠缓冲液(ph 6.0)配制0、50、100、200、400、600和1000μg/ml的葡萄糖溶液作为待测样品,用上述dns法处理后测定在540nm处(od
540
)的吸光度,并以葡糖糖浓度为横坐标、od
540
为纵坐标制作标准曲线。一个单位的β-1,3/4葡聚糖酶活性定义为上述反应条件下每分钟释放1.0μmol的还原糖(以葡萄糖为标准)所需的酶量。每次测定均设置3个平行。
173.薄层层析法检验水解产物:90μl含有10mm多糖底物的50mm乙酸钠缓冲液(ph 6.0)中加入10μl bbeng1蛋白(0.02m pbs溶解,2μg),在37℃孵育1h后,100℃5min终止反应。取10μl分5次点滴于提前110℃高温活化的硅胶板(德国默克)上,放于展层缸内,使用正丁醇-甲醇-水(8:4:3,v/v/v)作为溶剂体系,样品扩散至展层板4/5处时,取出展层板,自然晾干。将显色液(5%硫酸,0.5%香草醛溶于无水乙醇)喷洒在硅胶板上,然后在95℃下烘烤10min,使产物可视化。
174.液相色谱(hplc)确定底物特异性:90μl含有10mm多糖底物的50mm的乙酸钠缓冲液(ph 6.0)中加入10μl蛋白酶液(0.02m pbs溶解,2μg),在37℃孵育1h后,100℃,5min终止反应,最后使用0.22μm过滤头过滤,获得上样样品。采用p230ⅱ高效液相色谱系统,ri-201h示差折光检测器,s3100自动进样器(依利特,大连)。分析柱采用sugar-h型色谱柱(4.6mm
×
250mm,5m,月旭科技,上海),在80℃检测温度下,以5mm h2so4为流动相,流速0.6ml/min,上样量为20μl,进行检测(sugar-h使用说明书)。
175.结果表明,以大麦葡聚糖(barleyβ-glucan)为底物水解活性最高,比活力达到了12300u/mg。对酵母葡聚糖(yeast glucan)、茯苓聚糖(pachyma)具有较高水解活性,而对昆布多糖(laminarin)及纤维素钠(cmc-na)表现出较低程度的水解活性,但无法水解石耳葡聚糖(pustulan)和甲壳素(chitosan)右旋糖酐(dextran)(表1)。薄层层析(tlc)结果表明,
bbeng1水解酵母葡聚糖产生三糖(图11),而对其它多糖水解产物无法观测到,可能是产量过低。进一步采用液相色谱分析水解产物,结果显示bbeng1可水解的处理受试底物均产生新的同一类型产物(图11),表明bbeng1通过β-1,3/4糖苷键的水解改变了受试底物的结构完整性。根据bbeng1的底物水解活性,推断该酶为β-1,3/4-葡聚糖酶(ec 3.2.1.6)。
176.表1.bbeng1对不同多糖的降解活性
[0177][0178]
用dns法测定释放的还原糖含量。以活力最高的大麦β-葡聚糖为100%,其他底物的降解能力表示相对活性。
[0179]
8.bbeng1降解昆虫营养
[0180]
为探究bbeng1与昆虫营养利用的关系,利用纯化的bbeng1分别处理昆虫体壁、血腔成分及昆虫体内寡糖海藻糖,采用hplc检测降解产物。具体操作如下:
[0181]
取90μl的底物(10mg/ml昆虫体壁、100μl/ml的血淋巴和10mg/ml海藻糖,50mm的乙酸钠缓冲液,ph 6.0),加入10μl蛋白酶液(0.02m pbs溶解,2μg),在37℃孵育1h后,100℃、5min终止反应,最后使用0.22μm过滤头过滤,获得上样样品。液相色谱(hplc)确定底物特异性方法,操作同[实施例1]7。
[0182]
结果表明,dns法测定发现bbeng1处理昆虫营养底物后产生了新的小分子还原糖。进一步液相色谱检测表明,bbeng1水解昆虫体壁成分、1:10稀释的昆虫血淋巴产生了分子量较低的成分,而以海藻糖为底物未产生新的产物(图12)。
[0183]
9.超量表达bbeng1促进球孢白僵菌菌落生长
[0184]
为揭示bbeng1与球孢白僵菌生长发育的关系,分别在基础培养基(cza)、富营养培养基(sday)以及不同碳源培养基上比较研究野生型菌株(wt)、bbeng1敲除突变体(δbbeng1)、回复互补菌株(comp)和超量表达转化子bbeng1
oe-1、bbeng1
oe-2、bbeng1
oe-3(表达量分别提高2.17、7.25和235.55倍)的菌落生长。
[0185]
具体方法为:用0.05%(vol/vol)tween-80配制浓度为1
×
107孢子/ml分生孢子悬浮液,点滴法接种2μl至1/4sday(混合糖)、察氏培养基(czapek-dox agar)及以葡聚糖、海藻糖、葡萄糖、甘露糖苷、甘露糖、赤藻糖、半乳糖、果糖等量替换察氏培养基中蔗糖的培养基上,倒置于26℃培养箱,观察生长情况,测量培养3d至8d的菌落直径,以excel的slope函数计算菌落生长速率,采集第8d的菌落图片。
[0186]
结果表明,尽管球孢白僵菌在不同的培养基或不同营养条件下的生长速率存在差异,但bbeng1
oe
菌株生长速率均明显高于野生菌株(wt),且生长速率与基因表达倍数相一致,而突变体δbbeng1菌株的生长与野生菌株(wt)和回复互补转化子(comp)无显著差异
(图13)。以bbeng1
oe-2为例,分别以混合糖、蔗糖、葡聚糖、海藻糖、葡萄糖、甘露糖苷、甘露糖、赤藻糖、半乳糖、果糖为唯一碳源的培养基上,超量表达bbeng1菌株生长速率分别比wt提高了0.08、0.09、0.06、0.15、0.15、0.36、0.10、0.04和0.24倍(图13)。由此表明,超量表达bbeng1加速了球孢白僵菌的生长。
[0187]
9.bbeng1与球孢白僵菌分生孢子产生、形态和萌发的关系
[0188]
分生孢子产量参照zhang et al.(appl environ microbiol 2009,75:3787
–
3795)的方法进行。具体操作如下:在20ml czapek-dox agar(czapek)和1:4稀释的添加1%(wt/vol)酵母浸膏(yeast extract)sabouraud’s dextrose agar培养基(1/4sday)冷却至45℃,分别加入50μl 1
×
107孢子/ml的分生孢子悬浮液混匀,倒入直径为90mm的培养皿制备平板。平板于26℃和15h/9h的光照与黑暗交替循环条件下培养,在培养5d、10d和15d时,用直径1.0cm的打孔器在平板上打孔,每个平板三个菌饼,放入10ml离心管,添加6ml 0.05%(vol/vol)tween 80充分涡旋,然后用四层擦镜纸过滤除去菌丝碎片。用血球计数板于显微镜下计数分生孢子浓度,然后换算成培养基单位面积产生的分生孢子数。每个菌株设三个重复,每次试验重复三次。
[0189]
分生孢子大小测定:在显微镜下采用软件image-pro plus 6.0sorftware(bio-rad,usa)测定分生孢子长径和宽径,每个菌株至少测定400个孢子。
[0190]
检测结果发现,bbeng1破坏菌株在cza培养基上的分生孢子产量与野生型菌株无显著差异,而超量表达菌株(bbeng1
oe
)在cza培养基上培养10d和13d的分生孢子产量显著高于野生型菌株(提高0.36和0.63倍)。bbeng1破坏菌株在富营养的1/4sday培养基上的分生孢子产量比野生型菌株显著降低(降低14.67%)。超量表达菌株(bbeng1
oe
)在富营养的1/4sday培养基上培养时的分生孢子产量显著高于野生型菌株,培养5d、10d和15d分别比野生菌株提高了0.43、0.37和0.17倍(图13)。
[0191]
分子孢子大小测定结果发现,超量菌株(bbeng1
oe
)分生孢子明显膨大。统计数据表明,bbeng1
oe
菌株分生孢子横切面面积大于6μm2约占34.13%,而wt、δbbeng1、comp分别占5.47%、3.45%、9.53%。小于5μm2的分生孢子,超量bbeng1
oe
约占35.71%,而wt、δbbeng1菌株分别占57.85%和48.26%。由此表明,超量表达bbeng1菌株中膨大分生孢子的占比显著高于其它菌株(图13)。
[0192]
10.超量表达bbeng1提高了菌株萌发速率
[0193]
采用平板法检测各菌株分生孢子萌发速率。收集分生孢子培植浓度5
×
107个/ml孢子悬浮液,取100μl孢子悬浮液涂布接种于cza,26℃倒置暗培养,8h后每隔2h取样,点滴乳酸棉蓝染色后倒置于显微镜下,观察孢子萌发情况,统计萌发率。将芽管长度大于分生孢子直径时视为萌发,每个视野统计100个以上孢子,重复三次。采用graphad prism8绘制萌发曲线,以spss 17.0的probit分析计算萌发中时(gt
50
)。
[0194]
检测结果发现,超量菌株bbeng1
oe
萌发速率显著高于其它菌株(图13)。在基础盐培养基上,超量菌株萌发中时(gt
50
=9.03
±
0.06h)比野生型菌株(gt
50
=10.11
±
0.12h)缩短了1.07h(p《0.01),而基因敲除菌株萌发速率(gt
50
=10.34
±
0.22h)和回复菌株(gt
50
=10.18
±
0.11h)与野生型无明显差异。在富营养培养基(1/4sday)培养基上,bbeng1
oe
萌发速率(gt
50
=8.59
±
0.03h)也显著快于野生型菌株(gt
50
=9.84
±
0.10h),而基因敲除菌株(gt
50
=10.05
±
0.12h)和回复菌株(gt
50
=10.10
±
0.18h)与野生型无明显差异。
[0195]
11.超量表达bbeng1改变真菌细胞壁重塑
[0196]
细胞壁在维持细胞形状,控制细胞生长方面具有重要作用,为明确bbeng1对真菌自身的作用,从形态学研究了bbeng1对球孢白僵菌细胞壁的作用。观察菌株生长形态发现亲本菌株(wt)的菌落边缘菌丝较稀疏,菌丝分支较短,且菌丝均为向外延伸生长状态。而超量表达转化子(bbeng1
oe
)的菌落边缘菌丝致密且杂乱,菌丝分支更长且为弯曲状(图14)。接种分生孢子孢悬液,收集1/4sdb液体培养基培养12h的萌发菌丝,使用细胞壁几丁质特异性染料荧光增白剂(cfw)和细胞核染料(pi)染色,观察萌发菌丝形态、细胞壁、隔膜及核的分布情况。结果发现,相比于野生型菌株两端极性生长,超量菌株则长时间保持一端萌发和伸长(图14),数据统计发现超量菌株的两端极性生长率比野生菌株降低约54.6%(图14)。超量菌株(bbeng1
oe
)菌丝隔膜数量显著少于野生型菌株,核的数量无明显改变(图14)。
[0197]
透射电镜观察各菌株虫菌体、芽生孢子、分生孢子显微结构,发现破坏bbeng1菌株虫菌体细胞壁显著变薄(p《0.05),而超量表达菌株虫菌体细胞壁显著厚于野生型菌株,厚度提高约0.79倍(p《0.01)(图15)。bbeng1
oe
芽生孢子及分生孢子细胞壁厚度显著高于野生型,而δbbeng1这两种形态的细胞细胞壁厚度与野生型无明显差异(图15)。使用荧光增白剂cfw、葡聚糖特异性染料苯胺蓝分别对菌丝细胞壁几丁质、葡聚糖进行染色观察,并使用image j统计平均荧光值。结果表明,超量菌株(bbeng1
oe
)虫菌体细胞壁中几丁质成分显著低于野生型菌株,葡聚糖成分则显著高于野生型菌株,而敲除菌株(δbbeng1)几丁质含量显著高于野生型(p《0.01),但葡聚糖含量与野生型无明显差异(图15)。
[0198]
cfw染色方法:取1000μl的1
×
107细胞悬浮液离心弃上清,用1ml 0.1m pbs(ph 7.4)悬浮,然后加入1μl cfw染液(fluka),于26℃避光处理30min,用0.01m pbs清洗3次洗去未结合的染液,然后用0.1m pbs(ph 7.4)重悬细胞,于激光共聚焦显微镜观察,em=405nm。
[0199]
pi染色方法:取1000μl的1
×
107细胞悬浮液离心弃上清,用200μl 75%乙醇处理5min,1ml 0.1m pbs(ph 7.4)洗涤3次,然后加入10μl pi(碘化丙啶)工作液(终浓度50μg/ml),于26℃避光处理30min,用0.01m pbs清洗3次洗去未结合的染液,然后用0.1m pbs重悬分生孢子,于激光共聚焦显微镜观察,em=552nm。
[0200]
苯胺蓝染色方法:取1000μl的1
×
107细胞悬浮液离心弃上清,然后加入200μl苯胺蓝染液(终浓度50μg/ml,ph 9.5),于26℃避光处理30min,用0.01m pbs清洗3次洗去未结合的染液,然后用0.1m pbs重悬细胞,于激光共聚焦显微镜观察,em=405nm。
[0201]
透射电镜方法:收集wt、δbbeng1、bbeng1
oe
和comp的分生孢子、芽生孢子和虫菌体细胞,用0.1m pbs洗涤3次。将样品置于2.5%戊二醛(vol/vol)的0.1m pbs(ph 7.0)中,4℃固定过夜。固定后用0.1m pbs于4℃洗涤3次,每次15min;用1%(w/v)饿酸溶液固定样品1h,然后用0.1m pbs于4℃洗涤3次,依次用30%、50%、70%、80%和95%的乙醇溶液(vol/vol)进行脱水处理,每次15min,最后在100%乙醇溶液脱水1次,过渡到纯丙酮处理20min。脱水完成后,依次以1:1、1:3、0:1的丙酮:spurr包埋剂(vol/vol)各浸润2h,然后用100%包埋剂过夜浸润,样品置换至新的离心管,70℃加热过夜,获得包埋好的样品。用leica em uc7超薄切片机切75nm的切片,收集于镍网上。用醋酸双氧铀染色5min,柠檬酸铅染色3min,获得用于透射电镜的样品,于ht7800透射电子显微镜观察。
[0202]
12.超量表达bbeng1提高了菌株的毒力
[0203]
以大蜡螟三龄幼虫为试虫,采用“经典”体壁接种和微量注射接种两种方式进行生物测定。
[0204]“经典”体壁接种操作如下:收集1/4sday培养基上培养10d的分生孢子,使用0.05%(v/v)tween-80配制浓度为3
×
107孢子/ml分生孢子悬浮液,取1ml孢子悬浮液置于喷雾塔喷雾塔接种30头大蜡螟试虫,以0.05%(v/v)tween-80进行相同处理作为对照,每个处理重复三组。将处理的试虫置于培养皿放于26℃人工气候箱,使用无菌滤纸进行保湿,每隔12h添加定量无菌水润湿滤纸,接种48h后每隔12h统计一次昆虫死亡数。
[0205]
微量注射接种操作如下:配制菌株分生孢子孢悬液浓度为5
×
106孢子/ml,利用微量注射仪从试虫第2对腹足注射,接种剂量为2μl/虫,用0.05%(v/v)tween-80相同处理作为对照,每组30虫,三个重复。接种后置于26℃人工气候箱培养,接种24h后,每隔12h统计试虫的死亡数。
[0206]
生物测定试验重复三次,使用graphad prism8绘制kaplan-meyer生存曲线,采用log-rank检验分析组间差异,以spss 17.0的probit分析计算对昆虫的半致死时间(lt
50
)。
[0207]
生物测定结果显示,无论是体壁接种还是微量注射接种,超量表达菌株(bbeng1
oe
)毒力均显著提高,敲除菌株(δbbeng1)毒力显著降低。体壁接种法和微量注射接种法,野生型菌株(wt)处理试虫的半致死时间(lt
50
)分别为127.93
±
3.62h和89.28
±
2.67h,bbeng1
oe
处理的lt
50
分别比wt处理的lt
50
缩短了17.14h和14.16h(p《0.01),而δbbeng1处理的lt
50
则分别比wt处理的lt
50
滞后19.65h(p《0.01)和11.01h(p《0.05)(图16)。无论体壁浸染还是微量注射接种,wt、δbbeng1、bbeng1
oe
和comp菌丝均可从僵虫正常穿出生长和产孢(图16)。
[0208]
13.超量表达bbeng1影响虫菌体的发育分化
[0209]
为揭示破坏和超量表达bbeng1对菌体在虫体内的增殖及分化的影响,用qpcr方法测定了微量注射接种后不同时间虫菌体的数量,并使用显微镜观察虫菌体形态。
[0210]
虫菌体生物量测定:参考he et al.(environmental microbiology,2020,22(7):2514
–
2535)方法,以球孢白僵菌内参基因18s rrna为检测靶标,利用qpcr定量检测病原菌在昆虫体内的增殖量。标准曲线制作:接种球孢白僵菌野生型(wt)于1/4sdy液体培养基,26℃、200rpm培养4d,用4层滤纸过滤收集单细胞芽生孢子,血球计数板计数浓度,梯度稀释为1
×
108、5
×
107、2
×
107、1
×
107、2
×
106、1
×
106和2
×
105孢子/ml,各取200μl于4℃、5000rpm离心5min,收集芽生孢子,加入8μl细胞裂解液(0.3m naoh)裂解细胞,170μl中和液中和后获得扩增模板,以18s rrna-f/18s rrna-r为扩增引物进行实时荧光定量pcr扩增,获得相对定量值(relative quantity),以芽生孢子浓度为横坐标,相对定量值(relative quantity)为纵坐标绘制标准曲线(y=0.1636x-0.1338,r2=0.996)。
[0211]
虫菌体数量测定:收集在1/4sday营养培养基上培养10d的分生孢子,用0.05%tween-80(vol/vol)配制浓度为5
×
106孢子/ml悬浮液,微量注射2μl于大蜡螟幼虫第二对腹足,接种后36h、48h和60h收集昆虫血淋巴,每组选择10虫,每虫收集20μl,三组重复,冰上操作。4℃、5000rpm离心5min,收集沉淀,加入8μl细胞裂解液(0.3m naoh)裂解细胞,170μl中和液中和后获得扩增模板,以18s rrna-f/18s rrna-r为扩增引物进行实时荧光定量pcr获得相对定量值(relative quantity),对应标准曲线计算获得虫菌体数量。扩增体系如下:2
×
sybr buffer 5μl,5pmol/l引物各1μl,稀释的cdna链模板3μl。扩增程序如下:95℃2min;95℃5s,60℃30s,39个循环;65℃-95℃升高0.5℃/5s。
[0212]
虫菌体发育分化形态观察:收集在1/4sday营养培养基上培养10d的分生孢子,用0.05%tween-80(vol/vol)配制浓度为5
×
106孢子/ml孢子悬浮液,微量注射2μl于大蜡螟三龄幼虫第二对腹足,12h、24h、36h、48h和60h取昆虫血淋巴,制片后显微镜观察虫菌体形态。每组选择5头虫,三个重复。
[0213]
结果表明,bbeng1
oe
在虫体内的繁殖速度明显高于野生菌株(图17)。注射接种后36h-60h,bbeng1
oe
在虫体内的菌体繁殖数量比野生菌株提高了50.98%-74.7%(p《0.01,图17),而基因敲除菌株(δbbeng1)与野生型菌株无明显差异。镜检过程中发现,相对于野生菌株在虫体内形成短的或呈棍棒形的芽孢子,bbeng1
oe
在虫体内分化的菌体明显变长,有些菌体分化成具有分枝的长菌丝体(图17)。
[0214]
14超量表达bbeng1影响真菌逃避宿主免疫反应
[0215]
昆虫免疫反应主要包括体液免疫和细胞免疫两种,其中,细胞免疫则包括血细胞对病原菌的吞噬、包裹等作用,而体液免疫主要包括酚氧化酶级联反应产生黑色素包裹并清除病原微生物,toll途径产生抗菌肽,以及释放一些ros/rnos等分子抑制病原菌的生长和繁殖。因此,病原菌要成功定殖于宿主昆虫,需要克服昆虫免疫反应,利用昆虫体内营养物质,快速繁殖进而致死昆虫。采用观察昆虫体内黑化反应及虫菌体逃离血细胞包裹,注射接种大蜡螟后体腔内的酚氧化酶活性(po)活性及活性氧(ros)测定,注射接种后大蜡螟体腔抗菌肽相关基因表达模式分析等方法检测bbeng1与昆虫免疫防御的关系。具体操作方法如下:
[0216]
体表黑化结节数统计:采用体壁浸染和体腔微量注射两种方式进行。体壁浸染:收集在1/4sday营养培养基上培养10d的分生孢子,用0.9%的生理盐水配制浓度为3
×
107孢子/ml孢子悬浮液,取1ml喷雾塔均匀喷雾接种大蜡螟三龄幼虫,每处理30虫。微量注射:收集在1/4sday营养培养基上培养10d的分生孢子,用0.9%的生理盐水配制浓度为5
×
106孢子/ml孢子悬浮液,微量注射仪接种2μl于大蜡螟第二对腹足。24h、36h和48h时观察真菌感染过程中大蜡螟体表黑色结节变化,并拍照,统计数据。
[0217]
昆虫体内黑化反应、虫菌体逃离血细胞包裹:收集在1/4sday营养培养基上培养10d的分生孢子,使用0.9%的生理盐水配制浓度为5
×
106孢子/ml孢子悬浮液,微量注射2μl于大蜡螟第二对腹足,12h、24h、36h、48h和60h收集昆虫血淋巴,冰上操作,制片后显微镜观察黑化反应、虫菌体逃离血细胞包裹。每组选择10虫,三个重复。
[0218]
昆虫酚氧化酶(po)活性检测:参考yang et al.方法(appl environ microbiol 2014,78:5845
–
5854)进行。收集在1/4sday营养培养基上培养10d的分生孢子,用0.9%的生理盐水配制浓度为5
×
106孢子/ml孢子悬浮液,微量注射2μl于大蜡螟第二对腹足,接种后于0h、4h、8h、12h和24h收集昆虫血淋巴,冰上操作。每组选择10虫,每虫收集10μl于100μl ac buffer(anticoagulant solution(0.14m nacl,0.1m glucose,26mm citric acid,30mm trisodium citrate,10mm edta,ph 4.6))(wanchoo et al.,2009,microbiol-sgm,155:3121-3133),4℃、5000rpm离心5min,收集上清液。用0.1m磷酸缓冲液(ph 6.9)配置5mg/ml的多巴溶液(现配现用),取100μl上清液加入300μl多巴溶液,充分混匀。取100μl混合液加至酶标板中,酶标仪上0min测定一次490nm波长下的od值,作为od
490始
,孵育30min测定记录为od
490终
。每组样品重复三次,每分钟每δod
490
=0.01定义为一个单位的po活性(u),将od值转化为酶活单位。酶活性以活力单位u/ml表示,酶活=(od
490终-od
490始
)/0.01/30min/
虫血体积ml。
[0219]
活性氧(ros)测定:采用hydrogen peroxide colorimetric/fluorometric assay kit(biovision,usa)定量测定试剂盒测定昆虫血淋巴中的活性氧ros(h2o2)水平。具体如下:收集在1/4sday营养培养基上培养10d的分生孢子,用0.9%的生理盐水配制浓度为5
×
106孢子/ml孢子悬浮液,微量注射2μl于大蜡螟第二对腹足,接种后于0h、3h、6h、9h和12h收集昆虫血淋巴,冰上操作。每组选择15虫,每虫收集60μl,计900μl,于4℃、5000rpm离心5min。取上清750μl与50μl ac buffer及200μl浓度为10mg/ml的过氧化氢酶抑制剂(3-amino-1,2,4-triazole)混合,用0.22μm过滤头及10kda分子量截止自旋过滤器(康宁生命科学公司)过滤去除大分子杂质,获得待测液。取50μl待测液与50μl过氧化氢反应工作液混匀,避光孵育10min,酶标仪检测ex/em=535/587nm的荧光值,并根据标准曲线换算,最终获得昆虫血淋巴中的h2o2的浓度(pmol/μl)。
[0220]
抗菌肽相关基因表达模式测定:配制5
×
106孢子/ml孢子悬浮液,微量注射2μl于大蜡螟第二对腹足,接种后于12h和24h在石蜡盘上解剖昆虫收集脂肪体,采用easyspin植物rna快速提取试剂盒(北京艾德生物科技有限公司)提取rna,并参照试剂盒(oligo(dt)-primed cdna synthesis kit(mbi fermentas)说明书进行反转录获得cdna。以此cdna为模板,以大蜡螟肌动蛋白基因actin(gen-bank id:113519289)为参比基因,检测大蜡螟抗菌肽相关基因及toll途径相关基因的表达模。抗菌肽相关基因包括1个防御素蛋白(gal-p[ay528421])、3个抗真菌蛋白(glo[af394588.1]、cec[xm_026898304.2]、aap[di105103.1])、8个防御肽(mor-a[ef564370.1]、mor-b[ef564366.1]、mor-c[ef564365.1]、mor-d[ef564372.1]、mor-e[ef564369.1]、mor-f[ef564368.1]、mor-g[ef564367.1]和mor-h[af394588.1])和3个抗真菌肽(gal[af453824]、prp1[fj494919.1]和ap2[jq862476.1])基因。toll途径相关基因包括β-1,3glucan recognition protein gene(bgrp1,am265582.1)、(xm_031908808)和dorsal(xm_031907032)基因。引物序列如下:
[0221]
actin-f:5'-atctggcatcacaccttctacaacg-3'(seq id no.35)
[0222]
actin-r:5'-gacatacatagccggggagttgaag-3'(seq id no.36)
[0223]
cec-f:5'-atttgcctgcatcgtagcg-3'(seq id no.37)
[0224]
cec-r:5'-cttgtactgctggaccagctttt-3'(seq id no.38)
[0225]
gal-p-f:5'-gtggggtgcgacgaattaca-3'(seq id no.39)
[0226]
gal-p-r:5'-caagaagctgccgcaatgac-3'(seq id no.40)
[0227]
mor-a-f:5'-tgcccgttggtgccataaaa-3'(seq id no.41)
[0228]
mor-a-r:5'-ggctgtatacttcgtgcgct-3'(seq id no.42)
[0229]
mor-b-f:5'-tggtaaagctctgcgtggaa-3'(seq id no.43)
[0230]
mor-b-r:5'-tctttttcggtttgaactggct-3'(seq id no.44)
[0231]
mor-c-f:5'-aagcggcgcctaaagtcaat-3'(seq id no.45)
[0232]
mor-c-r:5'-ctgtactcgccgcactgatt-3'(seq id no.46)
[0233]
mor-d-f:5'-cgctctcaagaaaggcggaa-3'(seq id no.47)
[0234]
mor-d-r:5'-catgctcgtacacttgttggc-3'(seq id no.48)
[0235]
mor-e-f:5'-ttggcgccatcaagaaaggt-3'(seq id no.49)
[0236]
mor-e-r:5'-acgtggctgtaaacctcgtg-3'(seq id no.50)
[0237]
mor-f-f:5'-ctggtcaagccgaccctaag-3'(seq id no.51)
[0238]
mor-f-r:5'-ctgcctgttcctaacgtggt-3'(seq id no.52)
[0239]
mor-g-f:5'-gatgctcgccctgtttgttg-3'(seq id no.53)
[0240]
mor-g-r:5'-gcctgttcttgacgtggcta-3'(seq id no.54)
[0241]
mor-h-f:5'-cgttagcaagcagatgcacg-3'(seq id no.55)
[0242]
mor-h-r:5'-atttcgccatttctgccgac-3'(seq id no.56)
[0243]
ap2-f:5'-gtgcaaaatgcctttgactcg-3'(seq id no.57)
[0244]
ap2-r:5'-ttggcgcttctttcttctctgt-3'(seq id no.58)
[0245]
aap
–
f:5'-tccgttttgttgttggtctgc-3'(seq id no.59)
[0246]
aap
–
r:5'-cacacgcacctccctatcag-3'(seq id no.60)
[0247]
prp1-f:5'-ctaccgcatccatggtctcc-3'(seq id no.61)
[0248]
prp1-r:5'-ctttgccacggttgtgtacg-3'(seq id no.62)
[0249]
glo
–
f:5'-cgttagcaagcagatgcacg-3'(seq id no.63)
[0250]
glo
–
r:5'-atttcgccatttctgccgac-3'(seq id no.64)
[0251]
bgrp1-f:5'-agaatgccgactggtgactg-3'(seq id no.65)
[0252]
bgrp1-r:5'-ggatatgccatcaggcctcc-3'(seq id no.66)
[0253]-f:5'-tctgggccaacaacactagg-3'(seq id no.67)
[0254]-r:5'-accagtcagcgaagataccg-3'(seq id no.68)
[0255]
dorsal-f:5'-taaagcgcgatcgtacggag-3'(seq id no.69)
[0256]
dorsal-r:5'-ccgtgaagggatatgtgcgt-3(seq id no.70)
[0257]
结果表明,体壁侵染接种及微量注射接种发现,基因敲除菌株处理的昆虫表面出现更明显的黑化现象,而超量表达菌株处理则不产生明显黑化现象(图18)。进一步检测注射接种大蜡螟后体腔内的酚氧化酶活性及ros水平,发现敲除菌株处理的昆虫酚氧化酶活性显著高于野生菌株处理,ros水平也显著提高,而超量菌株处理的昆虫酚氧化酶活性及ros水平显著低于野生菌株处理(图18)。接种昆虫的抗菌肽基因表达水平分析发现,注射12h和24h后基因敲除菌株处理昆虫的抗菌肽相关基因及toll途径相关基因显著上调表达,而超量菌株处理的昆虫抗菌肽相关基因及toll途径相关基因表达量却显著低于野生型菌株(图18c)。以上结果表明bbeng1参与真菌逃避宿主免疫反应。
[0258]
15.超量表达bbeng1真菌细胞表面病原识别相关分子模式分布减少
[0259]
为明确bbeng1影响真菌逃避昆虫免疫的机制是否为改变了细胞表面病原识别相关分子模式分布,我们选择3种细胞凝集素和免疫荧光法检测虫菌体表面碳源表位和β-1,3-葡聚糖分布即病原相关分子模式分布,并采用image j_v1.8.0sorftware(national institutes of health,usa)统计荧光数值。
[0260]
碳源表位检测参考wannchoo et al.(microbiology 2009,155:3121-3133)方法,配制5
×
106孢子/ml孢子悬浮液,微量注射2μl至大蜡螟,48h收集血淋巴,4℃、10000rpm离心5min,弃去上清,使用0.1m pbs缓冲液洗涤5次。离心收集虫菌体细胞。配制不同凝集素反应体系(表2),加入虫菌体至终浓度为1
×
107细胞/ml,26℃避光过夜孵育。将处理好的细胞液离心,用0.1m pbs缓冲液洗涤5次洗去多余的染液,于激光共聚焦显微镜观察并拍照发射
波长em=488nm。
[0261]
表2配制不同凝集素反应体系
[0262]
名称简写反应缓冲液反应浓度共轭物(ex/em)(nm)concanavalin aconapbs,1.0mm cacl2,2.0mm mncl260μg/mlfluorescein(495/518)galanthus nivalis lectingnl10mm hepes,0.15m nacl,ph 7.520μg/mlfluorescein(495/518)wheatgerm agglutininwgapbs,1.0mm cacl220μg/mlfluorescein(495/518)
[0263]
细胞表面β-1,3-葡聚糖的检测:根据上述方法收集虫菌体,用4%的甲醛固定30min,然后用0.1m pbs缓冲液洗涤3次去除甲醛,获得固定好的虫菌体细胞。将虫菌体细胞重悬于包含0.1mg/ml的β-1,3-glucan抗体、1%tween-20(vol/vol)的0.1m pbs(ph=7.0),4℃避光过夜孵育。4℃、5000rpm离心5min,用预冷的0.1m pbs洗涤3次,将虫菌体细胞重悬于0.1m pbs稀释的fitc标记的二抗(终浓度为0.1mg/ml)中,室温避光孵育2h,离心去掉上清,再次使用0.1m pbs洗涤3次,于激光共聚焦显微镜观察并拍照,发射波长em=488nm。
[0264]
结果显示,敲除bbeng1虫菌体表面与凝集素cona、wga和gnl以及β-1,3-glucan抗体反应的荧光强度显著高于野生型菌株(wt),而超量表达菌株bbeng1
oe
虫菌体表面与cona、wga和β-1,3-glucan的荧光强度显著减弱,gnl荧光强度与野生菌株无无明显差异(图19)。由此表明,bbeng1影响真菌细胞表面特性,超量表达菌株bbeng1
oe
易逃避昆虫免疫识别及反应与其表面病原识别相关分子模式β-1,3-glucan、几丁质和甘露聚糖量显著减少相关。
[0265]
【实施例2】
[0266]
1.超量表达bbeng1的罗伯茨绿僵菌菌株构建
[0267]
利用组成型启动子pb3(seq id no.71)在罗伯茨绿僵菌中高水平表达球孢白僵菌bbeng1。
[0268]
构建超量bbeng1的罗伯茨绿僵菌表达载体策略如下:利用球孢白僵菌3-磷酸甘油醛脱氢酶基因的启动子pb3融合目的基因编码区序列,经遗传转化导入罗伯茨绿僵菌,组成型启动子pb3增加bbeng1的表达,利用rt-qpcr扩增筛选获得mr-bbeng1超量表达转化子。
[0269]
具体操作如下:
[0270]
以球孢白僵菌虫菌体cdna为模板,利用引物对oe-f2/oe-r2扩增bbeng1基因编码区(1767bp),将其克隆到pk2-pc-sur-tc-pb3(图2)的bamhⅰ和ecorv位点,置于组成型启动子pb3控制下,构建超量表达载体。bbeng1基因片段扩增体系如下:2
×
phanta max buffer 12.5μl,dntp mix 0.5μl,phanata max super-fidelity dna polymerase 0.5μl,5μmol/l引物oe-f2/oe-r2各1μl,球孢白僵菌虫菌体cdna 20ng,用水补足至25μl体系。扩增程序为:95℃5min;95℃30s,55℃30s,72℃2min,35个循环;72℃延伸10min。扩增产物于1.0%(w/v)的琼脂糖凝胶电泳,回收扩增片段测序验证。然后采用重组法将该片段连接于用bamhⅰ和ecorv酶切后的pk2-pc-sur-tc-pb3载体。重组法参考试剂盒(ii one step cloning kit c112(vazyme)说明书,形成超量表达bbeng1载体pk2-pc-sur-tc-pb3::bbeng1(图20)。
[0271]
利用农杆菌介导的真菌遗传遗传转化方法(ma et al.,2009,appl microbiol biotechnol 82:891
–
898)将表达载体pk2-pc-sur-tc-pb3::bbeng1转入罗伯茨绿僵菌野生型菌株,用氯磺隆除草剂抗性(4μg/ml)进行两次筛选获得转化子。用0.3m naoh裂解抗性转化子菌丝获得dna作为扩增模板,利用引物s5/s6为筛选引物扩增验证(530bp),筛选到多个
转化成功的转化子。
[0272]
验证后的转化子接种于1/4sdy液体培养基中,26℃、200rpm摇床培养3d,提取菌丝rna并反转录为cdna进行qrt-pcr分析筛选超量表达转化子。rna提取按照easyspin植物rna快速提取试剂盒(北京艾德生物科技有限公司)方法进行。紫外分光光度计定量rna。取1μg rna利用oligo(dt)引物反转录成cdna第一链,反转录参照试剂盒(oligo(dt)-primed cdna synthesis kit(mbi fermentas)说明书。将合成的cdna第一链稀释成10ng/μl,以罗伯茨绿僵菌三磷酸甘油醛脱氢酶基因mrgpd(gen-bank id:19261961)为参比基因,rt-qpcr检测bbeng1的转录。扩增体系如下:2
×
sybr buffer 5μl,5pmol/l引物各1μl,稀释的cdna链模板3μl。扩增程序如下:95℃2min;95℃5s,60℃30s,39个循环;65℃-95℃升高0.5℃/5s;扩增mrgpd和bbeng1转录本的引物对分别为mrgpd-f/mrgpd-r和rt1/rt2。结果表明,bbeng1在转化子中高水平转录,bbeng1相对转录水平为参比基因mrgpd的5.11-63.08倍(图21)
[0273]
oe-f2:5'-cccttttaatcaataacaggatccatgccgtcactcatttcgtg-3'(seq id no.72)
[0274]
oe-r2:5'-tcgacggtatcgataagcttgatatcttaggcacggcagatttggt-3'(seq id no.73)
[0275]
s5:5'-aatccgtgcccacgactacaa-3'(seq id no.74)
[0276]
s6:5'-cgaccatttggattggacgc-3'(seq id no.75)
[0277]
mrgpd-f:5'-gactgcccgcattgagaag-3'(seq id no.76)
[0278]
mrgpd-r:5'-gcttgacaaagttcttgttg-3'(seq id no.77)
[0279]
2.超量表达bbeng1加速了罗伯茨绿僵菌的生长
[0280]
为揭示超量表达bbeng1对罗伯茨绿僵菌生长的影响,检测了罗伯茨绿僵菌野生型菌株(wt)和超量表达转化子mr-bbeng1
oe
在固体培养基平板上的生长速率和液体培养基的生物量积累。固体平板生长速率测定的具体方法为:配制浓度为1
×
107个/ml孢子悬浮液,点滴法接种2μl于基础培养基(cza)、富营养培养基(pda),26℃恒温培养,测量第3-8d的菌落直径,利用最小二乘法计算日生长速率。液体培养基中菌株增殖测定:配制浓度为1
×
107个/ml孢子悬浮液,接种100μl孢子悬浮液至30ml pdb液体培养基,26℃、200rpm培养收集2d和4d收集菌体,烘干称量,计算生物量积累。
[0281]
结果表明,mr-bbeng1
oe
菌株在固体培养基上的生长速率均明显快于野生菌株(wt),在基础培养基(cza)和富营养培养基(pda)上分别比野生菌株提高0.02倍和0.06倍(p《0.01)(图22)。在液体培养基中,mr-bbeng1
oe
增殖也显著快于野生型菌株,培养2d和4d的mr-bbeng1
oe
生物量分别比野生型wt提高了4.37和1.91倍(p《0.01)。由此表明,超量表达bbeng1加速了罗伯茨绿僵菌的生长。
[0282]
3.超量表达bbeng1提高了罗伯茨绿僵菌分生孢子产量
[0283]
分生孢子产量参照zhang et al.(appl environ microbiol 2009,75:3787
–
3795)的方法进行。具体操作如下:在20ml czapek-dox agar(czapek)培养基冷却至45℃,分别加入50μl 1
×
107孢子/ml的分生孢子悬浮液混匀,倒入直径为90mm的培养皿制备平板。平板于26℃和15h/9h的光照与黑暗交替循环条件下培养,在培养5d、10d和15d时,用直径1.0cm的打孔器在平板上打孔,每个平板三个菌饼,放入10ml离心管,添加6ml 0.05%(vol/vol)tween 80充分涡旋,然后用四层擦镜纸过滤除去菌丝碎片。用血球计数板于显微
镜下计数分生孢子浓度,然后换算成培养基单位面积产生的分生孢子数。每个菌株设三个重复,每次试验重复三次。
[0284]
检测结果发现,超量表达bbeng1菌株(mr-bbeng1
oe
)在cza培养基上培养5d、10d和15d分生孢子产量比野生菌株分别提高了0.10、0.29和0.34倍(p《0.01)(图22)。由此表明,超量表达bbeng1促进了罗伯茨绿僵菌分生孢子产生。
[0285]
4.超量表达bbeng1提高了罗伯茨绿僵菌萌发速率
[0286]
采用平板法检测各菌株分生孢子萌发速率。收集分生孢子培植浓度5
×
107个/ml孢子悬浮液,取100μl孢子悬浮液涂布接种于cza,26℃倒置暗培养,4h后每隔2h取样,用乳酸棉蓝染色后倒置于显微镜下观察孢子萌发情况,统计萌发率,直至孢子萌发率达到95%以上。将芽管长度大于分生孢子直径时视为萌发,每个视野统计100个以上孢子,重复三次。采用graphad prism8绘制萌发曲线,以spss 17.0的probit分析计算萌发中时(gt
50
)。
[0287]
在基础培养基上,超量菌株(mr-bbeng1
oe
)萌发中时(gt
50
=6.33
±
0.15h)比野生型菌株(gt
50
=7.30
±
0.42h)提前0.97h(p《0.01)(图22)。由此表明,超量表达bbeng1显著加快了了罗伯茨绿僵菌萌发速率。
[0288]
5.超量表达bbeng1增强了罗伯茨绿僵菌的毒力
[0289]
以大蜡螟三龄幼虫为试虫,采用“经典”体壁接种和微量注射接种两种方式进行生物测定。
[0290]“经典”体壁接种操作如下:用0.05%(v/v)tween-80收集1/4sday培养基上培养10d的分生孢子,配制浓度为3
×
107孢子/ml分生孢子悬浮液,取1ml孢子悬浮液置于塔喷雾塔接种大蜡螟试虫,每处理90虫,分三组。以0.05%(v/v)tween-80进行相同处理作为对照。将处理的试虫置于培养皿放于26℃人工气候箱,用无菌滤纸保湿,接种48h后每隔12h统计昆虫死亡数。
[0291]
微量注射接种操作如下:配制菌株分生孢子孢悬液浓度为5
×
106孢子/ml,利用微量注射仪从试虫第2对腹足注射,接种剂量为2μl/虫,用0.05%(v/v)tween-80相同处理作为对照,每组30虫,分三组。接种后置于26℃人工气候箱培养,24h后每隔12h统计试虫的死亡数。
[0292]
生物测定试验重复三次,用graphad prism8绘制kaplan-meyer生存曲线,采用log-rank检验分析组间差异,以spss 17.0的probit分析计算对昆虫的半致死时间(lt
50
)。
[0293]
结果表明,无论是“经典”体壁接种还是微量注射接种,超量表达菌株(mr-bbeng1
oe
)毒力均显著提高(图23),体壁接种和微量注射接种超量表达菌株(mr-bbeng1
oe
)的半致死时间(lt
50
)比野生型菌株(wt)分别缩短了27.43h和4.19h(p《0.01)。由此表明,超量表达bbeng1显著提高了罗伯茨绿僵菌的毒力。
[0294]
【实施例3】
[0295]
1.超量表达bbeng1的蝗绿僵菌菌株构建
[0296]
利用组成型启动子pb3在蝗绿僵菌中高水平表达球孢白僵菌bbeng1。
[0297]
利用农杆菌介导的真菌遗传遗传转化方法(ma et al.,2009,appl microbiol biotechnol 82:891
–
898)将实施例2构建的超量表达bbeng1的载体pk2-sur-tc-pb3::bbeng1(图20)转入蝗绿僵菌野生型菌株,用氯磺隆除草剂抗性(4μg/ml)进行两次筛选获得转化子。0.3m naoh裂解抗性转化子菌丝获得dna作为扩增模板,利用引物s5/s6为筛选引物
扩增验证(530bp),筛选到多个转化成功的转化子。
[0298]
验证后的转化子接种于1/4sdy液体培养基中,26℃、200rpm摇床培养3d,提取菌丝rna并反转录为cdna进行qrt-pcr分析筛选超量表达转化子。rna提取按照easyspin植物rna快速提取试剂盒(北京艾德生物科技有限公司)方法进行。紫外分光光度计定量rna。取1μg rna利用oligo(dt)引物反转录成cdna第一链,反转录参照试剂盒(oligo(dt)-primed cdna synthesis kit(mbi fermentas)说明书。将合成的cdna第一链稀释成10ng/μl,以蝗绿僵菌的三磷酸甘油醛脱氢酶基因magpd(gen-bank id:19253895)为参比基因,rt-qpcr检测bbeng1的转录。扩增体系如下:2
×
sybr buffer 5μl,5pmol/l引物各1μl,稀释的cdna链模板3μl。扩增程序如下:95℃2min;95℃5s,60℃30s,39个循环;65℃-95℃升高0.5℃/5s;扩增magpd和bbeng1转录本的引物对分别为magpd-f/magpd-r和rt1/rt2。结果表明,bbeng1在转化子中高水平转录,bbeng1相对转录水平为参比基因mrgpd的1.38-73.25倍(图21)。
[0299]
magpd-f:5'-gactgcccgcattgagaag-3'(seq id no.78)
[0300]
magpd-r:5'-gcttgacaaagttcttgttg-3'(seq id no.79)
[0301]
2.超量表达bbeng1加速了蝗绿僵菌的生长
[0302]
为揭示超量表达bbeng1对蝗绿僵菌生长发育的影响,检测了野生型菌株(wt)和超量表达转化子ma-bbeng1
oe
在固体培养基平板上的生长速率和液体培养基的生物量积累。具体方法为,固体平板生长速率:配制浓度为1
×
107个/ml孢悬液,点滴法接种2μl基础培养基(cza)和富营养培养基(pda),26℃恒温培养,测量第3-8d的菌落直径。液体培养基测定生物量积累:接种100μl浓度为1
×
107个/ml孢子悬浮液于30ml pdb液体培养基,26℃、200rpm培养收集2d和4d收集菌体,烘干称量。
[0303]
结果表明,ma-bbeng1
oe
菌株固体培养基上生长速率均明显高于野生菌株(wt),在cza和pda上的生长速率分别比野生菌组提高了0.07倍和0.08倍(p《0.01)(图22)。ma-bbeng1
oe
在液体培养基中增殖也显著快于野生型菌株,培养2d和4d的生物量积累比野生型wt提高了1.05和0.42倍(p《0.01)(图22)。由此表明,超量表达bbeng1加速了蝗绿僵菌的生长。
[0304]
3.超量表达bbeng1提高了蝗绿僵菌分生孢子产量
[0305]
分生孢子产量参照zhang et al.(appl environ microbiol 2009,75:3787
–
3795)的方法进行。具体操作如下:在20ml czapek-dox agar(czapek)培养基冷却至45℃,分别加入50μl浓度为1
×
107孢子/ml分生孢子悬浮液,混匀后倒入直径为90mm的培养皿制备平板。平板于26℃和15h/9h的光照与黑暗交替循环条件下培养,在培养5d、10d和15d时,用直径1.0cm的打孔器在平板上打孔,放入10ml离心管,加入6ml 0.05%(vol/vol)tween 80充分涡旋,然后用四层擦镜纸过滤除去菌丝碎片。用血球计数板于显微镜下计数分生孢子浓度,然后换算成培养基单位面积产生的分生孢子数。每个菌株设三个重复,每次试验重复三次。
[0306]
检测结果发现,超量表达菌株(ma-bbeng1
oe
)在cza培养基上的分生孢子产量显著高于野生菌株,培养5d、10d及15d时分生孢子产量分别比野生菌株提高了0.11、0.87和0.50倍(p《0.01)(图22)。
[0307]
4.超量表达bbeng1增强了蝗绿僵的萌发速率
[0308]
采用平板法检测分生孢子萌发速率。取100μl浓度为5
×
107个/ml孢子悬浮液涂布
接种于cza,26℃倒置暗培养,8h后每隔2h取样,乳酸棉蓝染色后倒置于显微镜下观察孢子萌发情况,统计萌发率,直至孢子萌发率达到95%以上。将芽管长度大于分生孢子直径时视为萌发,每个视野统计100个以上孢子,重复三次。采用graphad prism8绘制萌发曲线,,以spss 17.0的probit分析计算萌发中时(gt
50
)。
[0309]
在基础培养基(cza)上,超量菌株(ma-bbeng1
oe
)萌发中时(gt
50
=10.15
±
0.14h)比野生型菌株(gt
50
=11.46
±
0.24h)提前1.32h(p《0.01)(图22)。由此表明,超量表达bbeng1促进了蝗绿僵菌萌发速率。
[0310]
5.超量表达bbeng1增强了蝗绿僵菌的毒力
[0311]
以五龄东亚飞蝗和三龄大蜡螟幼虫为试虫,分别采用体表浸染和体腔微量注射两种方法进行生物测定。
[0312]
体表浸染操作如下:收集1/4sday培养基培养10d的分生孢子,液体石蜡配制浓度为1
×
107个/ml孢子孢悬液,以液体石蜡为空白对照。每组分别吸取5μl孢子悬浮液点滴接种于飞蝗背板,每组点滴30虫。用新鲜的禾本科植物叶片喂养蝗虫,每隔12h统计蝗虫的死亡结果(注:蝗虫身体变红并呈僵硬状态即认为由蝗绿僵菌侵染致死),直至蝗虫全部死亡,每组实验重复三次以上。
[0313]
体内微量注射接种:收集1/4sday培养基培养10d的分生孢子,用0.05%(v/v)tween-80配制浓度为5
×
106孢子/ml的孢子悬浮液,用微量注射仪于三龄大蜡螟幼虫第2对腹足注射接种,2μl/虫,以0.05%(v/v)tween-80做相同处理作为对照,每组30虫,设三组。接种后置于26℃人工气候箱培养,接种24h后,每隔12h统计死亡数。
[0314]
生物测定试验重复三次,使用graphad prism8绘制kaplan-meyer生存曲线,采用log-rank检验分析组间差异,以spss 17.0的probit分析计算对昆虫的半致死时间(lt
50
)。
[0315]
结果表明,体壁接种五龄东亚飞蝗,超量表达菌株(ma-bbeng1
oe
)毒力显著高于野生亲本菌株(wt)(图23),半致死时间(lt
50
)比亲本菌株缩短了5.02h。微量注射大蜡螟幼虫结果显示,接种ma-bbeng1
oe
的致死速度显著快于亲本菌株(图23),半致死时间比亲本菌株处理缩短了和6.62h(p《0.01)。由此表明,超量表达bbeng1显著增强了蝗绿僵菌的毒力。
[0316]
【实施例4】
[0317]
1.mreng1基因克隆与序列分析
[0318]
以球孢白僵菌bbeng1氨基酸序列为探针,利用blastp搜索罗伯茨绿僵菌基因组(genbank:gca_000187405.1),克隆获得bbeng1同源蛋白编码基因mreng1(maa_09026,相似性为52.9%)(seq id no.80),基因组注释为concanavalin a-like lectin/glucanase。mreng1编码区(1487bp)含有3个内含子,编码一个含有432个氨基酸残基(48.1kda)的多肽。采用uniprot网站(https://www.uniprot.org/)中的blastp程序进行蛋白结构域分析显示,mreng1包含一个gh16(gh16_fungal_lam16a_glucanase)结构域,位于氨基端第39至275氨基酸。n端包括一个信号肽序列,无跨膜结构,无gpi锚定位点,含有多个糖基化位点,包含多个半胱氨酸残基,推测具有二硫键(图24)。
[0319]
2.mreng1在罗伯茨绿僵菌不同形态细胞中的表达模式分析
[0320]
为探究mreng1的表达模式,采用rt-qpcr分析mreng1在罗伯茨绿僵菌不同形态细胞的转录水平。具体操作如下:
[0321]
气生菌丝及分生孢子收集:用0.05%(v/v)的tween-80配制浓度为1
×
107孢子/ml
的罗伯茨绿僵菌野生型菌株分生孢子孢悬液,涂布法接种100μl至铺有玻璃纸的pdb固体培养基,26℃倒置培养3d收集气生菌丝。培养至10d,收集玻璃纸上菌体,0.05%(v/v)tween-80充分悬浮,用4层擦镜纸过滤除去菌丝,滤液离心,用无菌ddh2o洗涤2次,即为分生孢子样品。
[0322]
液生菌丝收集:配制浓度为1
×
107孢子/ml的罗伯茨绿僵菌野生型菌株分生孢子孢悬液,接种100μl至50ml pdb液体培养基,26℃、200rpm培养2d,去除培养液,固体沉淀即液生菌丝。
[0323]
虫菌体收集:配制浓度为1
×
107孢子/ml的罗伯茨绿僵菌野生型菌株分生孢子孢悬液,微量注射2μl至大蜡螟三龄幼虫,48h后收集试虫,液氮速冻。
[0324]
rna提取按照easyspin植物rna快速提取试剂盒(北京艾德生物科技有限公司)方法进行。紫外分光光度计定量rna。取1μg rna利用oligo(dt)引物反转录成cdna第一链,反转录参照试剂盒(oligo(dt)-primed cdna synthesis kit(mbi fermentas)说明书。将合成的cdna第一链稀释成10ng/μl,以18s rrna(gen-bank id:eu334679)为参比基因,rt-qpcr检测bbeng1的转录。扩增体系如下:2
×
sybr buffer 5μl,5pmol/l引物各1μl,稀释的cdna链模板3μl。扩增程序如下:95℃2min;95℃5s,60℃30s,39个循环;65℃-95℃升高0.5℃/5s。
[0325]
以mrgpd(genbank id:19261961)为参比基因,采用rt-qpcr检测mreng1的转录模式,扩增mrgpd和mreng1转录水平的引物对分别为mrgpd-f/mrgpd-r和rt3/rt4。结果表明,mreng1与bbeng1相似,仅在昆虫体内增殖的菌体-虫菌体高水平转录,而在腐生形态的细胞不转录(图25)。
[0326]
mrgpd-f:5'-gactgcccgcattgagaag-3'(seq id no.81)
[0327]
mrgpd-r:5'-gcttgacaaagttcttgttg-3'(seq id no.82)
[0328]
rt3:5'-ccacatccctgacgacaaca-3'(seq id no.83)
[0329]
rt4:5'-cgcaccatgtctatgcttgc-3'(seq id no.84)
[0330]
3.超量表达mreng1的罗伯茨绿僵菌构建
[0331]
利用组成型启动子pb3在罗伯茨绿僵菌中高水平表达mreng1。
[0332]
构建罗伯茨mreng1超量表达载体策略如下:利用球孢白僵菌3-磷酸甘油醛脱氢酶基因的启动子pb3融合目的基因编码区序列,经遗传转化导入罗伯茨绿僵菌,组成型启动子pb3增加mreng1的表达,利用rt-qpcr扩增筛选获得mr-mreng1超量表达转化子。
[0333]
具体操作如下:
[0334]
以罗伯茨绿僵菌基因组dna为模板,利用引物对oe-f3/oe-r3扩增mreng1基因编码区(1487bp),将其克隆到pk2-pc-sur-tc-pb3(图2)的bamhⅰ和ecorv位点,置于组成型启动子pb3控制下,构建超量表达载体。目的片段扩增体系如下:2
×
phanta max buffer 12.5μl,dntp mix 0.5μl,phanata max super-fidelity dna polymerase 0.5μl,5μmol/l引物oe-f3/oe-r3各1μl,罗伯茨绿僵菌基因组dna 20ng,用水补足至25μl体系。扩增程序为:95℃5min;95℃30s,55℃30s,72℃1min 30s,35个循环;72℃延伸10min。扩增产物在1.0%(w/v)的琼脂糖凝胶电泳,回收扩增片段测序验证。然后采用重组法将该片段连接于用bamhⅰ和ecorv酶切后的pk2-pc-sur-tc-pb3,重组法参考试剂盒(ii one step cloning kit c112(vazyme))说明书,形成超量表达载体pk2-pc-sur-tc-pb3::mreng1(图
80充分涡旋,然后用四层擦镜纸过滤除去菌丝碎片。用血球计数板于显微镜下计数分生孢子浓度,然后换算成培养基单位面积产生的分生孢子数。每个菌株设三个重复,每次试验重复三次。
[0346]
检测结果发现,超量表达菌株(mr-mreng1
oe
)在cza培养基上培养5d分生孢子产量显著低于野生型菌株49.7%,但培养至10d及15d时超量菌株分生孢子产量则显著高于野生型菌株,分别比野生菌株提高了0.12和0.24倍(p《0.01)(图22)。
[0347]
5.超量表达mreng1提高了罗伯茨绿僵菌萌发速率
[0348]
采用平板法检测各菌株分生孢子萌发速率。取100μl浓度为5
×
107个/ml孢子悬浮液涂布接种于cza,26℃倒置暗培养,4h后每隔2h取样,棉蓝染色后倒置于显微镜下观察孢子萌发情况,统计萌发率,直至孢子萌发率达到95%以上。将芽管长度大于分生孢子直径时视为萌发,每个视野统计100个以上孢子,重复三次。采用graphad prism8绘制萌发曲线,以spss 17.0的probit分析计算萌发中时(gt
50
)。
[0349]
在基础培养基上,超量菌株mr-mreng1
oe
萌发速率显著快于野生型菌株,萌发中时(gt50=6.59
±
0.25h)比野生型菌株(gt
50
=7.30
±
0.42h)缩短论了0.71h(p《0.01)(图22)。由此表明,超量表达mreng1促进了罗伯茨绿僵菌孢子萌发。
[0350]
6.超量表达mreng1增强了罗伯茨绿僵菌的毒力
[0351]
以三龄大蜡螟幼虫为试虫,采用“经典”体壁接种和微量注射接种两种方式进行生物测定。
[0352]“经典”体壁接种操作如下:收集1/4sday培养基上培养10d的分生孢子,用0.05%(v/v)tween-80配制浓度为3
×
107孢子/ml分生孢子悬浮液,取1ml孢子悬浮液置于喷雾塔喷雾接种大蜡螟试虫,每组30虫,处理三组,以0.05%(v/v)tween-80进行相同处理作为对照。将处理的试虫置于培养皿放于26℃人工气候箱,无菌滤纸进行保湿,接种48h后每隔12h统计一次昆虫死亡数。
[0353]
微量注射接种操作如下:配制菌株分生孢子孢悬液浓度为5
×
106孢子/ml,利用微量注射仪从试虫第2对腹足注射,接种剂量为2μl/虫,用0.05%(v/v)tween-80相同处理作为对照,每组30头,处理3组。接种后置于26℃人工气候箱培养,接种24h后,每隔12h统计试虫的死亡数。
[0354]
生物测定试验重复三次,使用graphad prism8绘制kaplan-meyer生存曲线,采用log-rank检验分析组间差异,以spss 17.0的probit分析计算对昆虫的半致死时间(lt
50
)。
[0355]
结果显示,无论是“经典”体壁接种还是微量注射接种,超量表达mreng1显著增强了菌株毒力(图23),接种超量表达菌株mr-mreng1
oe
的半致死时间(lt
50
)分别缩短了33.91h和2.30h(p《0.01)。由此表明,超量表达mreng1显著增强了罗伯茨绿僵菌的毒力。
[0356]
【实施例5】
[0357]
1.maeng1基因克隆及序列分析
[0358]
以球孢白僵菌bbeng1氨基酸序列为探针,利用blastp搜索蝗绿僵菌(genbank:gca_000187425.2)基因组,获得bbeng1同源蛋白maeng1(mac_06610,相似性55.6%)(seq id no.89),基因组注释为β-1,3-endoglucanase。maeng1编码区(1520bp)含有3个内含子,编码一个含有448个氨基酸残基(49.9kda)的多肽。采用uniprot网站(https://www.uniprot.org/)中的blastp程序进行蛋白结构域分析显示,maeng1包含一个gh16
synthesis kit,mbi fermentas)说明书。将合成的cdna第一链稀释成10ng/μl,以蝗绿僵菌三磷酸甘油醛脱氢酶基因magpd(gen-bank id:19253895)为参比基因,rt-qpcr检测maeng1的转录。扩增体系如下:2
×
sybr buffer 5μl,5pmol/l引物各1μl,稀释的cdna链模板3μl。扩增程序如下:95℃2min;95℃5s,60℃30s,39个循环;65℃-95℃升高0.5℃/5s。扩增magpd和maeng1转录本的引物对分别为magpd-f/magpd-r和rt5/rt6。结果表明,筛选到转化子maeng1转录水平比野生菌株提高了0.25-3.46倍(图21)。oe-f4:5'-cccttttaatcaataacaggatccatgcgccctgtaaccgcttg-3'
[0372]
(seq id no.94)
[0373]
oe-r4:5'-tcgacggtatcgataagcttgatatcttagatgtgtcgcgcaccat-3'
[0374]
(seq id no.95)
[0375]
s9:5'-aatccgtgcccacgactacaa-3'(seq id no.96)
[0376]
s10:5'-tgggtatccttttggccgtc-3'(seq id no.97)
[0377]
4.超量表达maeng1促进了蝗绿僵菌的生长
[0378]
为明确超量表达maeng1对蝗绿僵菌生长的影响,检测了野生型菌株(wt)和超量表达转化子ma-maeng1
oe
在固体培养基平板上的生长速率和液体培养基的生物量积累。具体方法为,固体平板生长速率测定:配制浓度为1
×
107个/ml孢子悬浮液,取2μl点滴接种于基础培养基(cza)和富营养培养基(pda),26℃恒温培养,测量第3-8d的菌落直径,计算日增长速率。液体培养基生物量积累测定:接种100μl浓度为1
×
107个/ml孢子悬浮液与30ml pdb液体培养基,26℃、200rpm培养,收集培养2d和4d菌丝,烘干至恒重,称量生物量。
[0379]
结果表明,超量表达maeng1促进了蝗绿僵菌在固体平板上的生化,在cza和pda上,ma-maeng1
oe
菌株的生长速率分别比野生菌株提高了0.10倍和0.07倍(p《0.01)(图22)。液体培养基中mr-mreng1
oe
增殖量也显著高于野生型菌株,培养2d和4d的生物量积累分别比野生型wt提高了0.33和0.16倍(p《0.01)(图22)。由此表明,超量表达maeng1促进了蝗绿僵菌的生长。
[0380]
5.超量表达maeng1提高了蝗绿僵菌分生孢子产量
[0381]
分生孢子产量参照zhang et al.(appl environ microbiol 2009,75:3787
–
3795)的方法进行。具体操作如下:在20ml czapek-dox agar(czapek)培养基冷却至45℃,加入50μl 1
×
107孢子/ml的分生孢子悬浮液混匀,倒入直径为90mm的培养皿制备平板。平板于26℃和15h/9h的光照与黑暗交替循环条件下培养,在培养5d、10d和15d时,用直径1.0cm的打孔器在平板上打孔,取菌饼放入10ml离心管,加入6ml 0.05%(vol/vol)tween 80充分涡旋,然后用四层擦镜纸过滤除去菌丝碎片。用血球计数板于显微镜下计数分生孢子浓度,然后换算成培养基单位面积产生的分生孢子数。每个菌株设三个重复,每次试验重复三次。
[0382]
检测结果发现,超量表达菌株(ma-maeng1
oe
)在cza培养基上培养5d时的分生孢子产量与野生型菌株无明显差异,但培养至10d和15d,分生孢子产量则显著高于野生型菌株,分别比野生菌株提高了0.94和0.34倍(p《0.01)(图22)。
[0383]
6.超量表达maeng1促进了蝗绿僵菌孢子萌发
[0384]
采用平板法检测各菌株分生孢子萌发速率。取100μl浓度为5
×
107个/ml孢子悬浮液涂布接种于cza,26℃倒置暗培养,8h后每隔2h取样,棉蓝染色后于倒置于显微镜下观察
孢子萌发情况,统计萌发率,直至孢子萌发率达到95%以上。将芽管长度大于分生孢子直径时视为萌发,每个视野统计100个以上孢子,重复三次。采用graphad prism8绘制萌发曲线,以spss 17.0的probit分析计算萌发中时(gt
50
)。
[0385]
结果表明,超量表达maeng1促进了蝗绿僵孢子萌发,其中超量表达菌株ma-maeng1
oe
萌发中时(gt
50
=.36
±
0.13h)比野生型菌株(gt
50
=11.46
±
0.24h)缩短了2.11h(p《0.01)(图22)。
[0386]
7.超量表达maeng1增强了蝗绿僵菌的毒力
[0387]
分别以东亚飞蝗五龄幼虫和大蜡螟三龄幼虫为试虫,采用体表侵染和体腔微量注射两种方法进行生物测定。
[0388]
体表侵染接种操作如下:收集1/4sday培养基培养10d的分生孢子,用液体石蜡配制浓度为1
×
107个/ml孢子孢悬液,以液体石蜡为空白对照。每组分别吸取5μl孢子悬浮液点滴于东亚飞蝗幼虫背板,每组点滴30虫,用新鲜的禾本科植物叶片喂养。每隔12h观察并统计蝗虫的死亡结果(注:蝗虫身体变红并呈僵硬状态即认为由蝗绿僵菌侵染致死),直至蝗虫全部死亡,每组实验重复三次以上。
[0389]
体内微量注射接种:收集1/4sday培养基培养10d的分生孢子,用0.05%(v/v)tween-80配制浓度为5
×
106孢子/ml的孢子悬浮液,采用微量注射仪注射接种大蜡螟三龄幼虫第2对腹足,2μl/虫,以0.05%(v/v)tween-80做相同处理作为对照,每组30虫,设置三组。接种后置于26℃人工气候箱培养,接种24h后,每隔12h统计试虫死亡数。
[0390]
生物测定试验重复三次,用graphad prism8绘制kaplan-meyer生存曲线,采用log-rank检验分析组间差异,以spss 17.0的probit分析计算对昆虫的半致死时间(lt
50
)。
[0391]
结果表明,超量表达maeng1显著增强了菌株毒力(图23)。体壁接种结果显示,超量表达菌株ma-maeng1
oe
对东亚飞蝗幼虫的半致死时间(lt
50
)比野生菌株缩短了17.14h,而微量注射接种后,ma-maeng1
oe
对大蜡螟幼虫的半致死时间(lt
50
)比野生菌株缩短了6.10h(p《0.01)。