1.本发明涉及帕唑帕尼三聚体杂质,具体涉及一种帕唑帕尼三聚体杂质中间体2,2'-((4-溴苯基)亚甲基)双(4-氯-1-甲基苯)的合成方法,属于药物化学技术领域。
背景技术:
2.帕唑帕尼是由葛兰素史克公司研发的一种可干扰顽固肿瘤存活和生长所需的新血管生成的新型口服血管生成抑制剂,可适用于晚期肾细胞癌、软组织肉瘤(sts)、上皮性卵巢癌和非小细胞肺癌(nsclc)的治疗。在帕唑帕尼的工业生产过程中会产生多种杂质,其中帕唑帕尼三聚体杂质分离过程繁琐、分子量大、疏水性强、分子高度对称不利于分离和分析,其结构式如下:
[0003][0004]
帕唑帕尼三聚体杂质的三芳基母核位阻较大,给该杂质的全合成带来了巨大的挑战。
技术实现要素:
[0005]
为了解决现有技术中的问题,本发明提供一种帕唑帕尼三聚体杂质中间体2,2'-((4-溴苯基)亚甲基)双(4-氯-1-甲基苯)(式4化合物)。
[0006]
除特殊说明外,本发明所述份数均为重量份,所述百分比均为质量百分比。
[0007]
为实现上述目的,本发明的技术方案为:
[0008]
第一方面,本发明提供式4化合物。式4化合物结构如下:
[0009]
[0010]
式4化合物可以作为原料或中间体合成帕唑帕尼三聚体杂质。
[0011]
本发明还提供上述式4化合物在制备帕唑帕尼三聚体杂质原料或中间体或杂质对照品中的用途。
[0012]
第二方面,本发明提供式3化合物。式3化合物结构如下:
[0013][0014]
本发明还提供上述式3化合物在制备帕唑帕尼三聚体杂质原料或中间体或式4化合物杂质对照品中的用途。
[0015]
式3化合物脱羟基还原得到式4化合物。脱羟基还原反应的溶剂包括二甲苯、乙酸乙酯、丙酮、乙腈、四氢呋喃、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二氯甲烷、乙醚、二氧六环中的一种或多种,脱羟基还原反应的还原剂包括氢气、二甲基二氯硅烷、碘化钠中的一种或多种。
[0016]
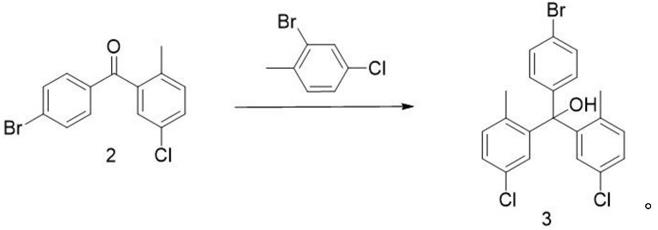
式3化合物由式2化合物与2-溴-4-氯甲苯经亲核反应制得,反应路线如下:
[0017][0018]
亲核反应的溶剂包括丙酮、四氢呋喃、乙醚、二氧六环中的一种或多种,所述亲核反应的催化剂包括镁、锌、异丙基氯化镁、异丙基溴化镁、异丙基锂、正丁基锂、异丁基锂、叔丁基锂中的一种或多种。
[0019]
式2化合物由式1化合物氧化得到,反应路线如下:
[0020][0021]
所述氧化反应的溶剂包括二氯甲烷、乙腈、丙酮、甲苯、四氢呋喃、乙醚、乙酸乙酯、二氧六环中的一种或多种,所述氧化反应的氧化剂包括臭氧、重铬酸钠、重铬酸钾、高锰酸钾、琼斯试剂、双氧水、间氯过氧苯甲酸、活性二氧化锰中的一种或多种。
[0022]
式1化合物由对溴苯甲醛与2-溴4-氯甲苯偶联得到,反应路线如下:
[0023][0024]
偶联反应的溶剂包括丙酮、四氢呋喃、乙醚、二氧六环中的一种或多种,所述偶联反应的引发剂包括碘、1,2-溴乙烷、1,2-氯乙烷中的一种或多种,所述偶联反应的催化剂为镁。
[0025]
有益效果:
[0026]
本发明提供一种帕唑帕尼三聚体杂质中间体(三芳基部分)的合成方法,经过四步反应,成功构建大位阻的三芳基化合物式(4)化合物。本发明以对溴苯甲醛为起始原料,与2-溴-4-氯甲苯偶联形成(4-溴苯基)(5-氯-2-甲苯基)甲醇,然后与氧化剂作用得到(4-溴苯基)(5-氯-2-甲苯基)甲酮,之后与2-溴-4-氯甲苯结合生成(4-溴苯基)双(5-氯-2-甲苯基)甲醇,然后与还原试剂反应,成功制得大位阻的2,2'-((4-溴苯基)亚甲基)双(4-氯-1-甲基苯)。本发明原料价廉易得,方法操作简单,适合工业化生产,可用于帕唑帕尼三聚体杂质的全合成。
具体实施方式
[0027]
下面通过具体实施例对本发明进行具体描述,在此指出以下实施例只用于对本发明进行进一步说明,不能理解为对本发明保护范围的限制,本领域的技术熟练人员可以根据上述发明内容对本发明作出一些非本质的改进和调整。本发明所用原料及试剂均为市售产品。
[0028]
实施例1
[0029]
化合物1的合成方法为:
[0030]
取100ml三口圆底烧瓶,往其中依次加入镁屑(0.65g,1eq),加入催化量的碘,四氢呋喃(10ml),在n2保护下缓慢滴加2-溴4-氯甲苯(4.44g,1eq)的四氢呋喃溶液(20ml),引发碘褪色后继续常温搅拌1~2h,然后向反应体系中缓慢滴加对溴苯甲醛(5g,1.25eq)的四氢呋喃溶液(10ml),加完后将反应升温至60℃反应1~8h,tlc点板监测反应完全,淬灭后搅拌30min,减压过滤,乙酸乙酯(20ml*3)萃取,无水硫酸钠干燥有机层后过滤,减压蒸干,过硅胶柱(洗脱剂石油醚比乙酸乙酯为40:1)得粗品,最后经石油醚乙酸乙酯(石油醚比乙酸乙酯=40:1)混合溶剂重结晶纯化制得白色固体4.04g,产率为60%。
[0031]
白色固体的表征数据如下:1hnmr(600mhz,cdcl3)δ7.17(d,j=8.4hz,2h),6.95(dd,j=8.1,2.2hz,1h),6.81(t,j=7.9hz,3h),5.39(s,1h),3.82(s,1h),1.85(s,3h).
13
c nmr(151mhz,cdcl3)δ142.64(s,1h),140.88(s,1h),133.61(s,1h),132.01(s,4h),131.72(s,5h),128.94(s,5h),127.70(s,3h),126.13(s,3h),121.89(s,1h),72.26(s,2h),18.84(s,3h).esi-ms[m+h]
+
=311.6.
[0032]
上述反应式为:
[0033][0034]
实施例2
[0035]
化合物2的合成方法为:
[0036]
取250ml单口圆底烧瓶,往其中加入化合物1(4g,36mmol),加入二氯甲烷15ml,搅拌溶解。0℃下滴加30ml琼斯试剂,反应24h,tlc点板监测反应完全,滴加10%氢氧化钠溶液调ph为中性,加水10ml,二氯甲烷(10ml*3)萃取,无水硫酸钠干燥有机层后过滤,减压蒸干,经硅胶柱(洗脱剂石油醚)得白色固体3.32g,产率为84%。白色固体的表征数据如下:1hnmr(600mhz,cdcl3)δ7.67
–
7.57(m,4h),7.35(d,j=8.1hz,1h),7.27(d,j=8.1hz,0h),7.22(d,j=8.2hz,1h),2.26(s,3h).
13
c nmr(151mhz,cdcl3copy)δ195.67(s,1h),139.42(s,1h),135.70(s,1h),135.01(s,1h),132.43(s,3h),131.92(s,5h),131.42(s,5h),131.18(s,1h),130.31(s,3h),128.82(s,1h),127.93(s,3h),19.32(s,2h).esi-ms[m+h]
+
=307.9.
[0037]
上述反应式为:
[0038][0039]
实施例3
[0040]
化合物3的合成方法为:
[0041]
取100ml三口圆底烧瓶,加入2-溴4-氯甲苯(0.53g,1eq),四氢呋喃30ml,-40℃n2保护下缓慢滴加n-buli(1.9ml,1.8eq),低温反应2h;加入化合物2(1g,1.25eq)的四氢呋喃(10ml)溶液,加毕,升温至45℃反应24h,tlc点板监测有反应,淬灭后搅拌30min,减压过滤,乙酸乙酯(20ml*3)萃取,无水硫酸钠干燥有机层后过滤,减压蒸干,经硅胶柱(洗脱剂石油醚比乙酸乙酯为40:1)纯化得白色固体0.68g,产率为61%。
[0042]
白色固体的表征数据如下:1hnmr(600mhz,dmso)δ7.59(d,j=8.8hz,1h),7.30(dd,j=8.1,2.3hz,1h),7.24(d,j=8.2hz,1h),7.17(d,j=8.3hz,1h),6.81(s,1h),6.68(d,j=2.2hz,1h),2.05(s,3h).
13
c nmr(151mhz,dmso)δ146.32(s,-1h),145.34(s,0h),137.07(s,-1h),134.63(s,1h),131.32(s,2h),130.53(s,3h),130.20(s,2h),128.21(s,3h),127.82(s,3h),121.05(s,-2h),82.33(s,-2h),21.92(s,-4h).esi-ms[m+h]
+
=433.9.
[0043]
上述反应式为:
[0044][0045]
参照上述实施例,发明人对化合物2经过2-溴-4-氯甲苯的亲核进攻制得化合物3的反应条件进行了探索,结果列于表1中。结果显示,当金属试剂分别使用mg,lda(二异丙基氨基锂)且其他条件不变时(表1,条目1,2),该反应没有目标化合物生成。当使用n-buli(正丁基锂)作为金属试剂时,其他条件不变,产物的收率为61%(表1,条目3)。此外,将化合物2加入到反应瓶过后,考察了升温对反应的影响,尝试了从-20℃-45℃不同温度下的反应,转化率升高,升高温度有利于反应的进行。具体来说,在-20℃反应目标产物收率痕量,将温度升高至45℃,可将目标产物收率达到中等(表1,条目3-6)。因此,将化合物2加入到反应瓶过后,将反应升温到45℃时可提高反应的收率(表1,条目3)。最终确定了反应的最佳条件,在-40℃下,以正丁基锂(1.8当量)为金属试剂,将化合物2加入到反应瓶过后,将反应升温到45℃在四氢呋喃中反应24h,反应收率最高,达到61%。
[0046]
表1反应条件筛选
[0047][0048]d痕量收率。
[0049]
实施例4
[0050]
化合物4的合成方法为:
[0051]
取100ml三口圆底烧瓶,依次加入加入化合物3(0.6g,1eq),ki(0.92g,4eq),乙腈(5ml),n2保护下逐渐滴加二甲基二氯硅烷(0.26ml,2eq),常温下反应12h,tlc点板监测反应完全,浓缩反应液,加入10ml水,乙酸乙酯(10ml*3)萃取,合并有机相,依次用饱和碳酸氢钠(10ml*2)、10%硫代硫酸钠(10ml*3)、饱和食盐水(10ml*2)洗涤,无水硫酸钠干燥有机层后过滤,减压蒸干,经硅胶柱(洗脱剂石油醚)纯化后得白色固体0.26g,产率为89%。
[0052]
白色固体的表征数据如下:1h nmr(600mhz,dmso)δ7.71
–
7.47(m,1h),7.27(d,j=1.7hz,2h),7.15
–
6.97(m,1h),6.55(s,1h),5.77(s,1h),2.10(s,3h).
13
c nmr(151mhz,dmso)δ143.38(s,2h),140.56(s,1h),135.98(s,2h),132.92(s,4h),132.12(d,j=8.3hz,8h),131.01(s,2h),128.20(s,4h),127.26(s,4h),120.65(s,1h),49.30(s,2h),18.98(s,4h).esi-ms[m+h]
+
=417.9.
[0053]
上述反应式为:
[0054]