1.本发明属于精细化工与专用化学品领域,具体为一种用于高表活体系的纤维素微凝胶基复配悬浮流变改性体系的制备方法。
背景技术:
2.以涂料、洗衣液为代表的高浓度表面活性剂产品,是现今人类社会发展不可或缺的生产、生活资料。表活产品浓缩化可以大幅度节约生产用水量以及包装和物流成本,符合国家节能减排发展战略的需求。近年来,功能性固体粒子在高表活体系中的悬浮问题,越来越引起人们的关注,例如在日化领域,洗涤产品的长效功能性(持久留香或长周期的抗霉、抗菌)均依赖于功能性微胶囊固体颗粒,但是,不难发现,市场上鲜有类似产品出现,其原因在于以微胶囊为代表的功能性固体颗粒难以在保持高表活体系产品原有粘度特性和配方的同时,稳定悬浮于表活产品中。
技术实现要素:
3.本发明提供一种用于高表活体系的纤维素微凝胶基复配悬浮流变改性剂的制备方法,以纤维素微凝胶为基质,通过自身纳米孔网络,协同可发生特异性相互作用的线性多糖聚合物构建“岛-链”状网络体系,在抵御高浓度表活分子强缔合作用的同时,提高流体的静态屈服性以及触变性;无毒天然可降解纤维素作为原料对其进行剪切分散后,使其具有良好的悬浮性,从而构建纤维素基微凝胶协同网络,实现在高表活体系中的运用。
4.本发明的技术方案如下:
5.一种用于高表活体系的纤维素微凝胶基复配悬浮流变改性剂的制备方法,对细菌纤维素进行改性后得到纤维素微凝胶,纤维素微凝胶再与线性多糖进行复配,得到纤维素微凝胶基复配悬浮流变改性剂。
6.所述细菌纤维素进行表面改性的方法包括阴离子改性、阳离子改性、醚化改性等。
7.所述细菌纤维素进行表面阴离子改性方法如下:将细菌纤维素、磷酸二氢钠溶液、tempo、次氯酸钠溶液、亚氯酸钠全部加入烧杯,盖住烧杯,在室温下500-600rpm搅拌4-5h,然后在60-65℃下缓慢搅拌48h,将悬浮液冷却至室温后,用去离子水过滤彻底洗涤tempo氧化的纤维素至ph值为中性,tds=0,4800r/min转速下搅拌分散20-30min,放入冰箱冷藏备用。
8.所述磷酸二氢钠溶液的浓度为0.05-0.1mmol/l,ph=6.5-7;次氯酸钠溶液的浓度为0.5-4mol/l;细菌纤维素、tempo、亚氯酸钠的质量比为1:0.064-0.128:4.52-36.16;细菌纤维素与磷酸二氢钠溶液的质量体积比g:ml为1:150-200,细菌纤维素与次氯酸钠溶液的质量体积比g:ml为1:7.5-15。
9.所述阳离子改性的方法如下:将细菌纤维素在4800r/min转速下分散处理15-30s,将分散后的细菌纤维素与质量分数20-25%的氢氧化钠溶液混合,室温下500-600rpm搅拌1-2h,然后加入2,3-环氧丙基三甲基氯化铵,室温下500-600rpm搅拌30-45min,再在60-65
℃下缓慢搅拌6-8h,将悬浮液冷却至室温后,用去离子水过滤彻底清洗至ph值为中性,tds=0,4800r/min分散20-30min,放入冰箱冷藏备用。
10.所述细菌纤维素与氢氧化钠溶液的质量比为1:100-150,细菌纤维素与2,3-环氧丙基三甲基氯化铵的质量比为1:3-4。
11.所述醚化改性的方法如下:将细菌纤维素在4800r/min转速下分散处理15-30s,将分散后的细菌纤维素与质量分数10-20%的氢氧化钠溶液混合,在室温下500-600rpm搅拌1-2h,然后加入环氧丙烷,在室温下500-600rpm搅拌30-45min,然后在60-65℃下缓慢搅拌6-8h,将悬浮液冷却至室温后,用去离子水过滤彻底清洗至是ph值为中性,tds=0,4800r/min分散20-30min,放入冰箱冷藏备用。
12.所述细菌纤维素与氢氧化钠溶液的质量比为1:100-150,细菌纤维素与环氧丙烷质量比为1:3-4。
13.本发明得到的纤维素微凝胶再与线性多糖进行复配,线性多糖包括但不限于羟丙基甲基纤维素、羧甲基纤维素、黄原胶、壳聚糖、结冷胶、卡拉胶、琼脂、瓜尔豆胶或藻酸盐等;纤维素微凝胶与线性多糖进行复配时,纤维素微凝胶与线性多糖的质量比为0.25:0.025-0.25。
14.本发明利用纤维素作为原料,主要是由于细菌纤维素与植物纤维素相比无木质素、果胶和半纤维素等伴生产物,具有高结晶度(可达95%,植物纤维素的为65%)和高的聚合度(dp值2000~8000),采用微生物发酵合成的多孔性网状纳米级生物高分子聚合物细菌纤维素作为原料对其进行改性之后,使其具有良好的悬浮性,从而构建纤维素基微凝胶协同网络,实现在高表活体系中的运用。
附图说明
15.图1是实施例1制备得到的微凝胶的sem图;
16.图2是实施例1制备得到的微凝胶的光学照片图;
17.图3是实施例1和对比例1产物的粒径分布图;
18.图4是aeo体系中小球悬浮性的测试图;
19.图5是aes体系中小球在悬浮性的测试图;
20.图6是ddac体系中小球在悬浮性的测试图;
21.图7为实施例3的产物的屈服值测定;
22.图8为实施例3与表活复配运用的屈服值测定;
23.图9为实施例12与表活复配运用的屈服值测定;
24.图10为实施例3的产物的触变环测定;
25.图11为实施例3与表活复配运用的触变环测定;
26.图12为实施例12与表活复配运用的触变环测定。
具体实施方式
27.下面结合附图对本发明做进一步详细描述,但应注意本发明的保护范围并不受这些实施例的限制,本发明实施例中使用的细菌纤维素为市场购买得到的糖蜜椰果(上海超欢食品有限公司),并采用去离子水将其清洗至洗液的导电率和去离子水一致和tds=0后
使用。
28.文中缩写:tempo:2,2,6,6-四甲基哌啶-氮-氧化物;tds:溶解性固体总量;microgel:纤维素微凝胶;cmc-na:羧甲基纤维素钠;nfc:纳米纤维素;aeo:月桂基聚氧乙烯酸;aes:月桂基聚氧乙烯硫酸酯钠;ddac:双癸基二甲基氯化铵;hpmc:羟丙基甲基纤维素。
29.实施例1
30.一种纤维素微凝胶的制备方法,具体步骤如下:
31.(1)配置浓度为0.05mmol/l的磷酸二氢钠缓冲溶液2000ml,用naoh溶液调节缓冲溶液ph=6.8;配置浓度为0.5mol/l的次氯酸钠溶液;
32.(2)按照细菌纤维素、tempo、亚氯酸钠的质量比为1:0.064:4.52的比例,用磷酸二氢钠缓冲溶液溶解tempo,加入含水率为99%的细菌纤维素,细菌纤维素与磷酸二氢钠溶液的质量体积比g:ml为1:200,加入次氯酸钠溶液,细菌纤维素与次氯酸钠溶液的质量体积比g:ml为1:7.5,加入亚氯酸钠,依次加入到烧杯中后,密封烧杯,在室温下550rpm搅拌4.5h,在62℃下500rpm搅拌48h,将悬浮液冷却至室温后,用去离子水洗涤至ph值为中性,tds=0,彻底洗涤tempo氧化的纤维素,得到纤维素微凝胶(记为microgel阴离子,下同),4800r/min分散25min,放入冰箱冷藏备用。
33.将所得产物按0.25wt%纤维素微凝胶(用0.25wt%microgel阴离子表示,下同)的比例稀释后进行测试。
34.实施例2
35.一种纤维素微凝胶的制备方法,具体步骤如下:
36.(1)配置浓度为0.08mmol/l的磷酸二氢钠缓冲溶液2000ml,用naoh溶液调节缓冲溶液ph=6.5;配置浓度为3mol/l的次氯酸钠溶液;
37.(2)按照细菌纤维素、tempo、亚氯酸钠的质量比为1∶0.128∶36.16的比例,用磷酸二氢钠缓冲溶液溶解tempo,加入含水率为99%的细菌纤维素,细菌纤维素与磷酸二氢钠溶液的质量体积比g∶ml为1∶150,加入次氯酸钠溶液,细菌纤维素与次氯酸钠溶液的质量体积比g∶ml为1∶15,加入亚氯酸钠,依次加入到烧杯中后,密封烧杯,在室温下500rpm搅拌5h,在60℃下500rpm搅拌48h,将悬浮液冷却至室温后,用去离子水洗涤至ph值为中性,tds=0,彻底洗涤tempo氧化的纤维素,得到纤维素微凝胶,4800r/min分散20min,放入冰箱冷藏备用。
38.将所得产物按0.25wt%microgel阴离子的比例稀释后进行测试。
39.实施例3
40.一种纤维素微凝胶的制备方法,具体步骤如下:
41.(1)配置浓度为0.1mmol/l的磷酸二氢钠缓冲溶液2000ml,用naoh溶液调节缓冲溶液ph=7;配置浓度为4mol/l的次氯酸钠溶液;
42.(2)按照细菌纤维素、tempo、亚氯酸钠的质量比为1∶0.1∶10的比例,用磷酸二氢钠缓冲溶液溶解tempo,加入含水率为99%的细菌纤维素,细菌纤维素与磷酸二氢钠溶液的质量体积比g∶ml为1∶180,加入次氯酸钠溶液,细菌纤维素与次氯酸钠溶液的质量体积比g∶ml为1∶10,加入亚氯酸钠,依次加入到烧杯中后,密封烧杯,在室温下600rpm搅拌4h,在65℃下500rpm搅拌48h,将悬浮液冷却至室温后,用去离子水洗涤至ph值为中性,tds=0,彻底洗涤tempo氧化的纤维素,得到纤维素微凝胶,4800r/min分散30min,放入冰箱冷藏备用。
43.将所得产物按0.25wt%microgel阴离子的比例稀释后进行测试。
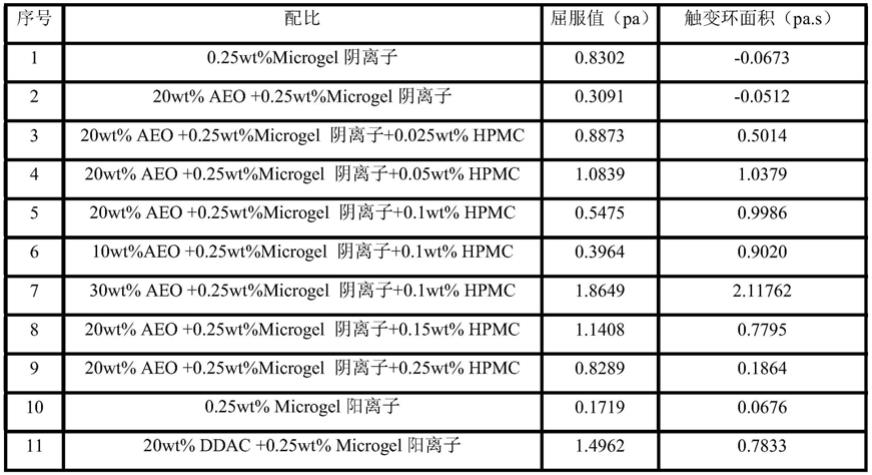
44.实施例3的产物按照下1稀释不同比例后进行测试,其屈服值如下表1所示。
45.表1
46.实施例序号microgel阴离子含量屈服值(pa)10.25wt%microgel阴离子0.830220.15wt%microgel阴离子0.308730.35wt%microgel阴离子1.183040.45wt%microgel阴离子1.6968
47.实施例4
48.一种纤维素微凝胶的制备方法,具体步骤如下:
49.将细菌纤维素在4800r/min转速下分散处理15s,将分散后的细菌纤维素与质量分数25%的氢氧化钠水溶液混合,细菌纤维素与氢氧化钠溶液的质量比为1∶120,在室温下550rpm搅拌1h,加入2,3-环氧丙基三甲基氯化铵,细菌纤维素与2,3-环氧丙基三甲基氯化铵的质量比为1:3.5,室温下600rpm搅拌30min,再在62℃下500rpm缓慢搅拌7h,将悬浮液冷却至室温后,用去离子水过滤清洗至ph为中性,tds=0,得到纤维素微凝胶(记为microgel阳离子,下同),4800r/min分散20min,放入冰箱冷藏备用。
50.将所得产物按0.25wt%microgel阳离子的比例进行稀释,进行流变学和悬浮性测试。
51.实施例5
52.一种纤维素微凝胶的制备方法,具体步骤如下:
53.将细菌纤维素在4800r/min转速下分散处理20s,将分散后的细菌纤维素与质量分数20%的氢氧化钠水溶液混合,细菌纤维素与氢氧化钠溶液的质量比为1:150,在室温下500rpm搅拌2h,加入2,3-环氧丙基三甲基氯化铵,细菌纤维素与2,3-环氧丙基三甲基氯化铵的质量比为1:4,室温下500rpm搅拌45min,再在60℃下500rpm缓慢搅拌8h,将悬浮液冷却至室温后,用去离子水过滤清洗至ph为中性,tds=0,4800r/min分散30min,放入冰箱冷藏备用。
54.将所得产物按0.25wt%microgel阳离子的比例进行稀释,进行流变学和悬浮性测试。
55.实施例6
56.一种纤维素微凝胶的制备方法,具体步骤如下:
57.将细菌纤维素在4800r/min转速下分散处理30s,将分散后的细菌纤维素与质量分数22%的氢氧化钠水溶液混合,细菌纤维素与氢氧化钠溶液的质量比为1:100,在室温下600rpm搅拌1.5h,加入2,3-环氧丙基三甲基氯化铵,细菌纤维素与2,3-环氧丙基三甲基氯化铵的质量比为1:3,室温下550rpm搅拌40min,再在65℃下500rpm缓慢搅拌6h,将悬浮液冷却至室温后,用去离子水过滤清洗至ph为中性,tds=0,4800r/min分散25min,放入冰箱冷藏备用。
58.将所得产物按0.25wt%microgel阳离子的比例进行稀释,进行流变学和悬浮性测试。
59.实施例7
60.一种纤维素微凝胶的制备方法,具体步骤如下:
61.将细菌纤维素在4800r/min转速下分散处理30s,将分散后的细菌纤维素与质量分数20%的氢氧化钠水溶液混合,在室温下600rpm搅拌1h,然后加入环氧丙烷,细菌纤维素与氢氧化钠溶液的质量比为1:100,细菌纤维素与环氧丙烷质量比为1:3,室温下550rpm搅拌40min,然后在65℃下500rpm缓慢搅拌7h,将悬浮液冷却至室温后,用去离子水过滤彻底清洗至ph值为中性,tds=0,得到纤维素微凝胶(记为microgel醚化,下同),4800r/min分散30min,放入冰箱冷藏备用。
62.将所得产物按0.25wt%microgel醚化的比例进行稀释,进行流变学和悬浮性测试。
63.实施例8
64.一种纤维素微凝胶的制备方法,具体步骤如下:
65.将细菌纤维素在4800r/min转速下分散处理15s,将分散后的细菌纤维素与质量分数10%的氢氧化钠水溶液混合,在室温下500rpm搅拌2h,然后加入环氧丙烷,细菌纤维素与氢氧化钠溶液的质量比为1:150,细菌纤维素与环氧丙烷质量比为1:4,室温下500rpm搅拌45min,然后在60℃下500rpm缓慢搅拌6h,将悬浮液冷却至室温后,用去离子水过滤彻底清洗至ph值为中性,tds=0,4800r/min分散20min,放入冰箱冷藏备用。
66.将所得产物按0.25wt%microgel醚化的比例进行稀释,进行流变学和悬浮性测试。
67.实施例9
68.一种纤维素微凝胶的制备方法,具体步骤如下:
69.将细菌纤维素在4800r/min转速下分散处理20s,将分散后的细菌纤维素与质量分数15%的氢氧化钠水溶液混合,在室温下550rpm搅拌1.5h,然后加入环氧丙烷,细菌纤维素与氢氧化钠溶液的质量比为1:120,细菌纤维素与环氧丙烷质量比为1:3.5,室温下600rpm搅拌30min,然后在62℃下500rpm缓慢搅拌8h,将悬浮液冷却至室温后,用去离子水过滤彻底清洗至ph值为中性,tds=0,4800r/min分散25min,放入冰箱冷藏备用。
70.将所得产物按0.25wt%microgel醚化的比例进行稀释,进行流变学和悬浮性测试。
71.实施例10
72.一种用于高表活体系的纤维素微凝胶基复配悬浮流变改性剂的制备方法,取实施例3制备得到的纤维素微凝胶再与线性多糖羟丙基甲基纤维素进行复配,纤维素微凝胶与线性多糖的质量比为0.25:0.025,得到纤维素微凝胶基复配悬浮流变改性剂。
73.实施例11
74.一种用于高表活体系的纤维素微凝胶基复配悬浮流变改性剂的制备方法,取实施例3制备得到的纤维素微凝胶再与线性多糖羟丙基甲基纤维素进行复配,纤维素微凝胶与线性多糖的质量比为0.25:0.05,得到纤维素微凝胶基复配悬浮流变改性剂。
75.实施例12
76.一种用于高表活体系的纤维素微凝胶基复配悬浮流变改性剂的制备方法,取实施例3制备得到的纤维素微凝胶再与线性多糖羟丙基甲基纤维素进行复配,纤维素微凝胶与线性多糖的质量比为0.25:0.1,得到纤维素微凝胶基复配悬浮流变改性剂。
77.实施例13
78.一种用于高表活体系的纤维素微凝胶基复配悬浮流变改性剂的制备方法,取实施例3制备得到的纤维素微凝胶再与线性多糖羟丙基甲基纤维素进行复配,纤维素微凝胶与线性多糖的质量比为0.25:0.15,得到纤维素微凝胶基复配悬浮流变改性剂。
79.实施例14
80.一种用于高表活体系的纤维素微凝胶基复配悬浮流变改性剂的制备方法,取实施例3制备得到的纤维素微凝胶再与线性多糖羟丙基甲基纤维素进行复配,纤维素微凝胶与线性多糖的质量比为0.25:0.25,得到纤维素微凝胶基复配悬浮流变改性剂。
81.实施例15
82.一种用于高表活体系的纤维素微凝胶基复配悬浮流变改性剂的制备方法,取实施例4制备得到的纤维素微凝胶再与线性多糖羧甲基纤维素进行复配,纤维素微凝胶与线性多糖的质量比为0.25:0.1,得到纤维素微凝胶基复配悬浮流变改性剂。
83.实施例16
84.一种用于高表活体系的纤维素微凝胶基复配悬浮流变改性剂的制备方法,取实施例7制备得到的纤维素微凝胶再与线性多糖壳聚糖进行复配,纤维素微凝胶与线性多糖的质量比为0.25:0.1,得到纤维素微凝胶基复配悬浮流变改性剂。
85.将上述实施例产物分别与活性剂进行复配使用,检测结果见下表2,配比中余量为水:
86.表2
[0087][0088][0089]
对比例1
[0090]
将细菌纤维素机械分散(4800r/min,15s),将所得产物按0.25wt%microgel的比例进行稀释,标记为0.25wt%机械,进行流变学和悬浮性测试。
[0091]
对比例2
[0092]
(1)按照质量比纤维素:h2o=1:100的比例,将1g纤维素溶解在100ml水中;
[0093]
(2)按照质量比纤维素:tempo=1:0.016的比例,取0.016g tempo配制tempo溶液
150ml;
[0094]
(3)按照质量比纤维素:溴化钾=1:0.1的比例,取0.1g溴化钾配制溴化钾溶液100ml;
[0095]
(4)按照质量比纤维素:naclo溶液的质量体积比g/ml为1:6.25,量取质量分数10%的naclo溶液,并将naclo溶液用0.1mol/l的hcl调节ph=10;
[0096]
(5)按比例将步骤(2)、(3)、(4)的溶液加入到步骤(1)的溶液中,室温下500rpm搅拌;
[0097]
(6)向步骤(5)中持续加入0.5mol/l的naoh溶液,维持ph值在9.8-10.2之间,直至未观察到naoh消耗;
[0098]
(7)将步骤(6)所得产物过滤洗涤,至ph值为中性,放入冰箱冷藏备用;
[0099]
(8)将步骤(7)所得产物按0.25wt%nfc的比例进行稀释,进行流变学和悬浮性测试。
[0100]
对比例3
[0101]
配制0.25wt%cnc-na溶液,标记为0.25wt%cnc-na,进行流变学和悬浮性测试。
[0102]
对比例4
[0103]
取对比例1制备得到的产物与线性多糖羟丙基甲基纤维素进行复配,产物与线性多糖羟丙基甲基纤维素的质量比为0.25:0.1,得到改性剂。
[0104]
对比例5
[0105]
取对比例2制备得到的产物与线性多糖羟丙基甲基纤维素进行复配,产物与线性多糖羟丙基甲基纤维素的质量比为0.25:0.1,得到变改性剂。
[0106]
对比例6
[0107]
取cnc-na与线性多糖羟丙基甲基纤维素进行复配,cnc-na与线性多糖的质量比为0.25:0.1,得到改性剂。
[0108]
将对比例得到产物分别与活性剂进行复配使用,检测其性能及使用效果,具体见下表3,配比中余量为水。
[0109]
表3
[0110]
测试序号配比屈服值(pa)触变环面积(pa.s)10.25wt%机械1.74790.8413220wt%aeo+0.25wt%机械1.50710.2985320wt%aeo+0.25wt%机械+0.1wt%hpmc1.09130.2343420wt%ddac+0.25wt%机械+0.1wt%hpmc1.60020.141350.25wt%nfc0.0656-0.5079620wt%aeo+0.25wt%nfc0.0837-0.2790720wt%aeo+0.25wt%nfc+0.1wt%hpmc0.1681-0.334380.25wt%cnc-na
‑‑
920wt%aeo+0.25wt%cnc-na-0.16661020wt%aeo+0.25wt%cnc-na+0.1wt%hpmc-0.1432
[0111]
对比表2和表3屈服力与触变环面积的数据可知,虽然单纯机械分散后的细菌纤维素的屈服值高于实施例1进行改性后的纤维素微凝胶,但是在浓度配比相同的情况下,单纯
机械分散后的细菌纤维素的触变环面积不如表2中纤维素微凝胶的好,而且单纯机械分散后的细菌纤维素在三种表活体系中运用时,它的触变环面积均减小,不符合该发明的目的,在与表活体系复配后的实施例屈服力和触变环面积均有所增加,说明实施例所制备得到的改性剂在高表活体系中能够起到提高悬浮固体颗粒在体系中稳定的效果,对比例1得到的单纯机械分散后的细菌纤维素则没有这种效果,对比例2纳米纤维素和对比例3得到的羧甲基纤维素钠本身没有或仅有极小的屈服值和触变环面积,在高表活体系中虽然会出现一点触变环,但效果仍然不明显,与线性多糖复配后效果也不甚理想,综上可知,实施例中制备得到的改性剂在表活体系中的效果明显。
[0112]
图1是实施例1制备得到的微凝胶的sem图;从图中可以看出所制备的微凝胶在不同倍数下的具体微观纤维形态。
[0113]
图2是实施例1制备得到的微凝胶的光学照片图;从图中可以看出所制备的微凝胶的颗粒大小及颗粒形状。
[0114]
图3是实施例1和对比例1制备得到的产物及不同表面活性剂复配之后的粒径分布图,从图中可以看出高速剪切预处理得到的细菌纤维素平均粒径为18.9um,已经达到微米级,经阴离子改性后的细菌纤维素在高速剪切后平均粒径为35.62um,两者相比,经阴离子改性后的细菌纤维素粒径分布更集中,体系颗粒大小更均匀,而高速剪切预处理得到的细菌纤维素虽然平均粒径较小,但体系颗粒大小没有阴离子改性后的均匀。
[0115]
图4、图5、图6是对小球在悬浮性的测试图,其中图4是aeo体系中小球悬浮性的测试图:aeo(a1、a2、a3)、aeo+microgel阴离子(b1、b2、b3)、aeo+卡波姆941(c1、c2、c3)在常温、4℃、和45℃下的悬浮性测试,图5是aes体系中小球在悬浮性的测试图:aes(a1、a2、a3)、aes+microgel阴离子(b1、b2、b3)、aes+卡波姆941(c1、c2、c3)在常温、4℃、和45℃下的悬浮性测试;图6是ddac体系中小球在悬浮性的测试图:ddac(a1、a2、a3)、ddac+microgel阴离子(b1、b2、b3)、ddac+卡波姆941(c1、c2、c3)在常温、4℃、和45℃下的悬浮性测试;从图中可以看出,在aeo体系中加入microgel阴离子和卡波姆941对小球都有较稳定的悬浮性,在aes体系和ddac体系中,microgel阴离子仍然能保持对小球的悬浮性,而卡波姆在ddac中已经无法存在。
[0116]
图7、图8、图9是实施例3和实施例12制备得到的产物与表活体系aeo复配得到的流变屈服值图;其中图7为实施例3的产物、图8为实施例3与表活复配的运用、图9为实施例12与表活复配的运用,从图中可以看出图7屈服值为0.8302pa,图8屈服值为0.3091pa,图9屈服值为0.5475pa,由此可见,aeo与microgel阴离子复配时,两者相互缠结,从而降低了体系的屈服值,而在加入hpmc后,屈服值有所提高。
[0117]
图10、图11、图12是实施例3和实施例12制备得到的产物与表活体系aeo复配得到的流变触变环图,其中图10为实施例3的产物、图11为实施例3与表活复配的运用、图12为实施例12与表活复配的运用,从图中可以看图10的触变环面积为-0.0673,图11的触变环面积为-0.0512,图12的触变环面积为0.9986,由此可见,aeo体系与microgel阴离子复配后,可以提高体系的悬浮性,而在加入hpmc后,体系的触变环面积更是大幅提高,触变环面积的变大表明在体系中构成触变网络的连接点增多,在进行剪切时,需要耗费的能量更高,同时也反映出体系的弹性在增强,这与静态屈服力增大的实验结果相对应。
[0118]
表活在单独使用时,没有屈服值,也没有触变环,说明单纯的表活没有办法稳定悬
浮固体粒子,在加入改性后的纤维素微凝胶后,体系中会产生屈服值,在与线性多糖复配后,产生屈服值和触变环,小球悬浮测试也说明单纯的表活不能让小球悬浮,在加入改性后的纤维素微凝胶与线性多糖复配后,可以让小球悬浮,而且在三种表活体系中都可以稳定悬浮固体粒子,但是卡波姆只在aeo体系中可以悬浮固体粒子,说明表活与纤维素微凝胶和线性多糖复配后,悬浮效果优于单纯的表活及常规改性剂卡波姆。