一种n-(n-三氟甲基亚胺酰基)吡啶盐的制备方法及应用
技术领域
1.本发明涉及有机合成技术领域,具体涉及一种n-(n-三氟甲基亚胺酰基)吡啶盐的制备方法及应用及其制备方法和应用。
背景技术:
2.n-亚胺酰基吡啶盐是一种重要的腈鎓离子衍生物,该种吡啶稳定的腈鎓离子通常利用原位形成的策略广泛应用于合成脒、酯、硫代酰胺、噻唑啉、炔基亚胺等化合物。由于稳定性较差,分离并运用n-(n-烷基/芳基亚胺酰基)吡啶盐的相关工作并不常见,而关于n-(n-三氟甲基亚胺酰基)吡啶盐则未见报道。将三氟甲基引入有机分子往往对其理化性质或生物活性产生深远的影响,其中向n原子上引入三氟甲基所形成的化合物在医药、农用化学品以及材料科学等领域有非常重要的应用,然而由于n-三氟甲基化方法学的匮乏,限制了含ncf3化合物的应用。鉴于n-亚胺酰基吡啶盐在合成含氮化合物应用方面的广泛性,开发n-(n-三氟甲基亚胺酰基)吡啶盐的合成方法及其在含ncf3化合物的合成应用具有重要研究价值。
3.在药物分子中,n-cf3唑是一类有价值的优势结构。例如,n-cf3唑类化合物相对于n-ch3唑类化合物具有更优异的生物活性。但由于合成方法的匮乏,导致应用方面发展缓慢。目前报道的n-cf3唑类化合物合成方法有如下三种:1、对唑类化合物杂环n上的官能团进行氟化转换;2、2011年togni课题组发展的唑类化合物杂环n上的原位n-硅烷化-脱硅三氟甲基化反应;3、2016年beier课题组发展的使用tmscf3与tsn3在低温下制备cf3n3的四氢呋喃溶液,并在铜催化条件下与炔烃发生[3+2]环加成反应。上述三种方法都存在明显的缺点如:需要在唑类化合物杂环n上进行繁琐的预官能化、需要过量的昂贵或毒性较大的氟化试剂、缺乏原子经济性及步骤经济性,对大量生产或环境友好极为不利、产率较低、底物普适性较差。因此发明一种简单温和高效的方法合成n-cf3唑类化合物在有机氟化学中具有非常迫切的需要。
技术实现要素:
[0004]
为解决上述技术问题,本发明提供一种n-(n-三氟甲基亚胺酰基)吡啶盐及其制备方法和应用;向腈类分子中加入phicf3cl,加热反应后,向反应体系中加入4-二甲胺基吡啶反应得到所述n-(n-三氟甲基亚胺酰基)吡啶盐。
[0005]
本发明的技术方案之一,一种n-(n-三氟甲基亚胺酰基)吡啶盐,化学结构式如式(1)所示:
[0006][0007]
进一步地,所述的芳基为苯基、带有给电子取代基的芳基、带有吸电子取代基的芳
基中的一种,所述的杂芳基为含硫杂环、含氧杂环、含氮杂环中的一种。
[0008]
本发明的技术方案之二,一种上述n-(n-三氟甲基亚胺酰基)吡啶盐的制备方法,向腈类化合物中加入phicf3cl,加热反应后,向反应体系中加入4-二甲胺基吡啶(dmap)反应得到所述n-(n-三氟甲基亚胺酰基)吡啶盐。
[0009]
进一步地,制备过程在氮气氛围下进行;所述腈类化合物为烷基腈或芳基腈或杂芳基腈;所述加热反应温度为25~100℃,加热反应时间为1~12h;phicf3cl在腈类化合物中的物质的量浓度为0.3~1.0mmol/ml;dmap与phicf3cl的摩尔比为1:1.5~1:3。
[0010]
本发明的技术方案之三,上述n-(n-三氟甲基亚胺酰基)吡啶盐在制备n-cf3化合物中的应用,所述n-cf3化合物为n-cf3脒和n-(n-cf3亚胺酰基)n杂环化合物、n-cf3亚胺酸酯、n-cf3硫代亚胺酸酯、n-cf3四氮唑、n-cf3咪唑和n-cf3三氮唑类化合物中的任意一种;
[0011]
根据产物不同,具体反应如下:
[0012]
反应
①
:将所述n-(n-三氟甲基亚胺酰基)吡啶盐与胺反应制备n-cf3脒;或,将所述n-(n-cf3亚胺酰基)dmap盐与n杂环化合物反应制备n-(n-cf3亚胺酰基)n杂环;
[0013]
反应
②
:将所述n-(n-三氟甲基亚胺酰基)吡啶盐与醇或酚反应制备n-cf3亚胺酸酯;
[0014]
反应
③
:将所述n-(n-三氟甲基亚胺酰基)吡啶盐与硫醇或硫酚反应制备n-cf3硫代亚胺酸酯;
[0015]
反应
④
:将所述n-(n-三氟甲基亚胺酰基)吡啶盐与叠氮化合物反应制备n-cf3四氮唑;
[0016]
反应
⑤
:将所述n-(n-三氟甲基亚胺酰基)吡啶盐与异氰化合物反应制备n-cf3咪唑;
[0017]
反应
⑥
:将所述n-(n-三氟甲基亚胺酰基)吡啶盐与重氮化合物反应制备n-cf3三氮唑。
[0018]
进一步地,所述反应
①
、反应
②
和反应
③
中,以三乙胺作为碱,以四氢呋喃或二氯甲烷作为溶剂;三乙胺的加入量为胺或n杂环化合物或醇或酚或硫醇或硫酚的物质的量的1~2倍,胺或n杂环化合物或醇或酚或硫醇或硫酚在溶液中的物质的量浓度为0.13~0.4mmol/ml,n-(n-三氟甲基亚胺酰基)吡啶盐在溶液中的物质的量浓度为0.13~0.3mmol/ml。
[0019]
进一步地,所述反应
④
、反应
⑤
和反应
⑥
中,以三乙胺或dbu作为碱,以二氯甲烷或dmf作为溶剂;三乙胺或dbu的加入量为叠氮化合物或亚甲基异氰或重氮化合物的物质的量的1~2倍量,叠氮化合物或亚甲基异氰或重氮化合物在溶液中的物质的量浓度为0.13~0.4mmol/ml,n-(n-三氟甲基亚胺酰基)吡啶盐和叠氮化合物或异氰化合物或重氮化合物的物质的量比为2.0:1~1.2:1,n-(n-三氟甲基亚胺酰基)吡啶盐在溶液中的物质的量浓度为0.1~0.24mmol/ml。
[0020]
进一步地,所述反应
①
中胺为脂肪族胺类化合物,所述n杂环化合物为吲哚、吡唑、咪唑或1,2,4-三氮唑,所述反应
②
中醇或酚为脂肪醇或苯酚类化合物;所述反应
③
中硫醇或硫酚为脂肪硫醇或苯硫酚类化合物;所述反应
④
中叠氮化合物为nan3;所述反应
⑤
中异氰基化合物为含有活泼亚甲基的异氰化合物;所述反应
⑥
中重氮化合物为重氮甲烷类化合物。
[0021]
本发明的技术方案之四,一种n-cf3化合物,具体为n-cf3脒、n-(n-cf3亚胺酰基)n杂环化合物、n-cf3亚胺酸酯、n-cf3硫代亚胺酸酯、n-cf3四氮唑、n-cf3咪唑和n-cf3三氮唑类化合物中的任意一种;所述n-cf3化合物由上述n-(n-三氟甲基亚胺酰基)吡啶盐制备。
[0022]
与现有技术相比,本发明具有以下有益效果:
[0023]
本发明首次合成了n-(n-三氟甲基亚胺酰基)吡啶盐,并利用其作为n-三氟甲基亚胺酰基合成子,与胺反应得到n-cf3脒、与n杂环化合物反应得到n-(n-cf3亚胺酰基)n杂环、与醇或酚反应得到n-cf3亚胺酸酯、与硫醇或硫酚反应n-cf3硫代亚胺酸酯,产率都非常高。而且本发明制备的n-(n-三氟甲基亚胺酰基)吡啶盐还可作为一类亲偶极体,与nan3或亚甲基异氰或重氮甲烷类化合物等1,3-偶极子进行反应,在室温下即可高效的得到n-cf3四氮唑、n-cf3咪唑、n-cf3三氮唑类化合物,与现有技术相比,所使用的化学试剂简单易得、反应条件更温和、产率更高、底物普适性更宽泛。
附图说明
[0024]
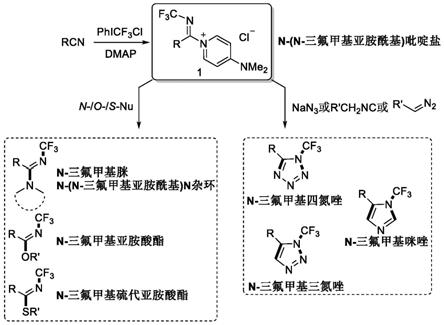
图1为本发明的技术路线图;
[0025]
图2为本发明实施例1制备的n-(n-cf3亚胺酰基)吡啶盐1的氢谱图;
[0026]
图3为本发明实施例1制备的n-(n-cf3亚胺酰基)吡啶盐1的碳谱图;
[0027]
图4为本发明实施例1制备的n-(n-cf3亚胺酰基)吡啶盐1的氟谱图;
[0028]
图5为本发明实施例2制备的n-cf3脒1的氢谱图;
[0029]
图6为本发明实施例2制备的n-cf3脒1的碳谱图;
[0030]
图7为本发明实施例2制备的n-cf3脒1的氟谱图;
[0031]
图8为本发明实施例3制备的n-(n-cf3亚胺酰基)n杂环1的氢谱图;
[0032]
图9为本发明实施例3制备的n-(n-cf3亚胺酰基)n杂环1的碳谱图;
[0033]
图10为本发明实施例3制备的n-(n-cf3亚胺酰基)n杂环1的氟谱图;
[0034]
图11为本发明实施例4制备的n-cf3亚胺酸酯1的氢谱图;
[0035]
图12为本发明实施例4制备的n-cf3亚胺酸酯1的碳谱图;
[0036]
图13为本发明实施例4制备的n-cf3亚胺酸酯1的氟谱图;
[0037]
图14为本发明实施例4制备的n-cf3亚胺酸酯2的氢谱图;
[0038]
图15为本发明实施例4制备的n-cf3亚胺酸酯2的碳谱图;
[0039]
图16为本发明实施例4制备的n-cf3亚胺酸酯2的氟谱图;
[0040]
图17为本发明实施例4制备的n-cf3硫代亚胺酸酯1的氢谱图;
[0041]
图18为本发明实施例4制备的n-cf3硫代亚胺酸酯1的碳谱图;
[0042]
图19为本发明实施例4制备的n-cf3硫代亚胺酸酯1的氟谱图;
[0043]
图20为本发明实施例4制备的n-cf3硫代亚胺酸酯2的氢谱图;
[0044]
图21为本发明实施例4制备的n-cf3硫代亚胺酸酯2的碳谱图;
[0045]
图22为本发明实施例4制备的n-cf3硫代亚胺酸酯2的氟谱图;
[0046]
图23为本发明实施例5制备的n-cf3四氮唑1的氢谱图;
[0047]
图24为本发明实施例5制备的n-cf3四氮唑1的碳谱图;
[0048]
图25为本发明实施例5制备的n-cf3四氮唑1的氟谱图;
[0049]
图26为本发明实施例6制备的n-cf3咪唑1的氢谱图;
[0050]
图27为本发明实施例6制备的n-cf3咪唑1的碳谱图;
[0051]
图28为本发明实施例6制备的n-cf3咪唑1的氟谱图;
[0052]
图29为本发明实施例7制备的n-cf3三氮唑1的氢谱图;
[0053]
图30为本发明实施例7制备的n-cf3三氮唑1的碳谱图;
[0054]
图31为本发明实施例7制备的n-cf3三氮唑1的氟谱图。
具体实施方式
[0055]
现详细说明本发明的多种示例性实施方式,该详细说明不应认为是对本发明的限制,而应理解为是对本发明的某些方面、特性和实施方案的更详细的描述。
[0056]
应理解本发明中所述的术语仅仅是为描述特别的实施方式,并非用于限制本发明。另外,对于本发明中的数值范围,应理解为还具体公开了该范围的上限和下限之间的每个中间值。在任何陈述值或陈述范围内的中间值以及任何其他陈述值或在所述范围内的中间值之间的每个较小的范围也包括在本发明内。这些较小范围的上限和下限可独立地包括或排除在范围内。
[0057]
除非另有说明,否则本文使用的所有技术和科学术语具有本发明所述领域的常规技术人员通常理解的相同含义。虽然本发明仅描述了优选的方法和材料,但是在本发明的实施或测试中也可以使用与本文所述相似或等同的任何方法和材料。本说明书中提到的所有文献通过引用并入,用以公开和描述与所述文献相关的方法和/或材料。在与任何并入的文献冲突时,以本说明书的内容为准。
[0058]
在不背离本发明的范围或精神的情况下,可对本发明说明书的具体实施方式做多种改进和变化,这对本领域技术人员而言是显而易见的。由本发明的说明书得到的其他实施方式对技术人员而言是显而易见得的。本技术说明书和实施例仅是示例性的。
[0059]
关于本文中所使用的“包含”、“包括”、“具有”、“含有”等等,均为开放性的用语,即意指包含但不限于。
[0060]
图1为本发明的技术路线图。
[0061]
实施例1:(n-(n-三氟甲基亚胺酰基)吡啶盐的制备)
[0062]
氮气氛围下,向10ml schlenk封管中加入腈分子(0.5ml),phicf3cl(93mg,0.3mmol)和搅拌子,反应在80℃搅拌2h后冷却至室温,然后加入dmap(24.4mg,0.2mmol)和二氯甲烷(0.5ml),氮气条件下继续搅拌1h。反应混合物用二氯甲烷稀释(20ml),减压浓缩后加入20ml石油醚,析出的白色固体用乙醚洗涤,得到n-(n-三氟甲基亚胺酰基)吡啶盐。
[0063][0064]
具体的,芳基腈为苯甲腈时,获得产物产率70%。其核磁和质谱表征:1h nmr(400mhz,cdcl3):δ=3.60(s,6h),7.45(d,j=8.0hz,2h),7.51(d,j=7.6hz,2h),7.63(t,j=7.8hz,2h),7.72(t,j=7.4hz,1h),8.36(d,j=8.0hz,2h).
13
c nmr(151mhz,cdcl3):δ=41.8(2c),109.0(2c),121.6(q,j=261.6hz),126.5,127.9(2c),129.2(2c),132.7,
137.1(2c),158.1,162.9(q,j=6.8hz).
19
f nmr(470mhz,cdcl3):δ=-52.3(s,3f).hrms(esi-tof)calc’d for c
15h15
f3n3[m-cl]
+
:294.1213;found 294.1217。产物的氢谱见图2,碳谱图见图3,氟谱图见图4。
[0065]
更换原料,可获得其他产物和产率如下:
[0066][0067]
实施例2:(n-cf3脒的合成)
[0068]
氮气氛围下,向10ml schlenk封管中加入n-(n-三氟甲基亚胺酰基)吡啶盐(72.6mg,0.22mmol),胺类化合物(0.2mmol),三乙胺(0.4mmol,56μl),四氢呋喃(1.5ml)和搅拌子,反应在室温搅拌10min后,反应混合物中加15ml水淬灭反应,用二氯甲烷(20ml
×
3)萃取,有机相用浓盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩,硅胶柱层析分离提纯(石油醚/乙酸乙酯=5:1)得到n-cf3脒类化合物。具体反应如下:
[0069][0070]
具体的,胺为二乙胺时,获得产物产率82%,其核磁和质谱表征:1h nmr(600mhz,cdcl3):δ=0.99(t,j=7.2hz,3h),1.28(t,j=7.2hz,3h),2.98(q,j=7.2hz,2h),3.63(q,j=7.0hz,2h),7.22(d,j=7.8hz,2h),7.38
–
7.42(m,3h).
13
c nmr(151mhz,cdcl3):δ=12.0,14.1,41.5,43.6,124.7(q,j=252.2hz),127.1(2c),128.1(2c),129.1,133.3,165.6(q,j=7.3hz).
19
f nmr(565mhz,cdcl3):δ=-43.6(s,3f).hrms(esi-tof)calc’d for c
12h15
f3n2na[m+na]
+
:267.1080;found 267.1082。产物的氢谱见图5,碳谱图见图6,氟谱图见图7;
[0071]
更换胺类化合物,可获得其他产物,具体结构和产率如下:
[0072][0073]
实施例3:(n-(n-cf3亚胺酰基)n杂环的合成)
[0074]
氮气氛围下,向10ml schlenk封管中加入n-(n-三氟甲基亚胺酰基)吡啶盐(0.22mmol),n杂环化合物(0.2mmol),三乙胺(0.4mmol,56μl),四氢呋喃(1.5ml)和搅拌子,反应在室温搅拌20min后,反应混合物中加15ml水淬灭反应,用二氯甲烷(20ml
×
3)萃取,有机相用浓盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩,硅胶柱层析分离提纯(石油醚/乙酸乙酯=5:1)得到n-(n-cf3亚胺酰基)n杂环。具体反应如下:
[0075][0076]
r为苯基,n杂环化合物为苯并咪唑时,获得产物产率92%,其核磁和质谱表征:1h nmr(600mhz,cdcl3):δ=7.42(dd,j1=6.6hz,j2=4.2hz,2h),7.49(d,j=9.0hz,2h),7.58(t,j=9.3hz,2h),7.63
–
7.67(m,2h),7.75
–
7.82(m,1h),8.24(s,1h).
13
c nmr(125mhz,cdcl3):δ=116.7,120.7,123.2(q,j=258.0hz),125.5,125.8,127.8(2c),128.9(2c),130.5,131.6,132.0,143.2,144.8,162.0(q,j=7.6hz).
19
f nmr(470mhz,cdcl3):δ=-50.4(s,3f).hrms(esi-tof)calc’d for c
15h11
f3n3[m+h]
+
:290.0900;found 290.0904。产物的氢谱见图8,碳谱图见图9,氟谱图见图10。
[0077]
更换n杂环化合物,可获得其他产物,具体结构和产率如下:
[0078][0079]
实施例4:(n-cf3亚胺酸酯或硫代亚胺酸酯的合成)
[0080]
氮气氛围下,向10ml schlenk封管中加入n-(n-三氟甲基亚胺酰基)吡啶盐(0.22mmol),醇或酚或硫醇或硫酚类化合物(0.2mmol),三乙胺(0.4mmol,56μl),二氯甲烷(1.5ml)和搅拌子,反应在室温搅拌20min后,反应混合物中加15ml水淬灭反应,用二氯甲烷(20ml
×
3)萃取,有机相用浓盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩,硅胶柱层析分离提纯(石油醚/乙酸乙酯=30:1)得到n-cf3亚胺酸酯或硫代亚胺酸酯。具体反应如下:
[0081][0082]
具体的,r为苯基,醇为苄醇时,获得产物产率89%,其核磁和质谱表征:1h nmr(600mhz,cdcl3):δ=5.35(s,2h),7.34(t,j=7.2hz,1h),7.36
–
7.43(m,6h),7.48(t,j=7.5hz,1h),7.52(d,j=7.8hz,2h).
13
c nmr(151mhz,cdcl3):δ=70.3,123.2(q,j=255.9hz),127.8(d,j=1.5hz,2c),128.2(2c),128.4(2c),128.5,128.6(2c),131.2,132.0,135.1,169.3(q,j=7.0hz).
19
f nmr(470mhz,cdcl3):δ=-48.5(s,3f).hrms(esi-tof)calc’d for c
15h12
f3nnao[m+na]
+
:302.0763;found 302.0762。产物的氢谱见图11,碳谱图见图12,氟谱图见图13;
[0083]
更换原料可以获得其他产物,具体结构和产率如下:c15h12
f3nnas[m+na]
+
:318.0535;found 318.0541。产物的氢谱见图17,碳谱图见图18,氟谱图见图19。
[0089]
r为苯基,硫酚为苯硫酚时,获得产物产率93%。其核磁和质谱表征:1h nmr(500mhz,cdcl3):δ=7.02
–
7.05(m,3h),7.10
–
7.14(m,2h),7.28
–
7.35(m,5h).
13
c nmr(125mhz,cdcl3):δ=122.0(q,j=260.8hz),126.8(2c),128.2(2c),129.0,129.1(2c),129.9,130.7,134.8(2c),135.7,183.9(q,j=7.0hz).
19
f nmr(470mhz,cdcl3):δ=-50.4(s,3f).for z-isomer,1h nmr(500mhz,cdcl3):δ=7.02
–
7.05(m,2h),7.10
–
7.14(m,1h),7.28
–
7.35(m,5h),7.40
–
7.46(m,2h).
13
c nmr(125mhz,cdcl3):δ=122.9(q,j=261.5hz),127.9(2c),128.1,129.2(4c),130.3,130.9,134.1(2c),136.5,179.9(q,j=8.0hz).
19
f nmr(470mhz,cdcl3):δ=-56.8(s,3f).hrms(esi-tof)calc’d for c
14h10
f3nnas[m+na]
+
:304.0378;found 304.0376。产物的氢谱见图20,碳谱图见图21,氟谱图见图22;
[0090]
更换原料可获得其他产物,具体结构和产率如下:
[0091][0092]
实施例5:(n-cf3四氮唑的合成)
[0093]
氮气氛围下,向10ml schlenk封管中加入n-(n-cf3亚胺酰基)dmap盐(0.24mmol),叠氮化钠(13mg,0.2mmol),二氯甲烷(1.5ml)和搅拌子,反应在室温搅拌30min后,用二氯甲烷稀释后,过滤,减压浓缩,硅胶柱层析分离提纯(石油醚/乙酸乙酯=50:1)得到n-cf3四氮唑。具体反应如下:
[0094][0095]
具体的,腈为苯甲腈时,获得产物产率92%,其核磁和质谱表征:1h nmr
(600mhz,cdcl3):δ=7.59(t,j=7.8hz,2h),7.67(t,j=7.8hz,1h),7.74(d,j=7.2hz,2h).
13
c nmr(151mhz,cdcl3):δ=117.4(q,j=271.4hz),121.8,129.1(2c),129.2(2c),132.5,154.0.
19
f nmr(470mhz,cdcl3):δ=-54.8(s,3f).hrms(esi-tof)calc’d for c8h6f3n4[m+h]
+
:215.0539;found 215.0533。产物的氢谱见图23,碳谱图见图24,氟谱图见图25;
[0096]
更换原料,可获得其他产物和产率如下:
[0097][0098]
实施例6:(n-cf3咪唑的合成)
[0099]
氮气氛围下,向10ml schlenk封管中加入n-(n-三氟甲基亚胺酰基)吡啶盐(0.22mmol),异氰化合物(0.2mmol),三乙胺(0.4mmol,56μl),dmf(4ml)和搅拌子,反应在室温搅拌12h后,反应混合物中加15ml水淬灭反应,用二氯甲烷(20ml
×
4)萃取,有机相用浓盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩,硅胶柱层析分离提纯(石油醚/乙酸乙酯=5:1)得到n-cf3咪唑类化合物。具体反应如下:
[0100][0101]
具体的,腈为苯甲腈,异氰化合物为异氰乙酸乙酯时,获得产物产率87%,其核磁和质谱表征:1h nmr(600mhz,cdcl3):δ=1.17(t,j=7.2hz,3h),4.22(q,j=7.0hz,2h),7.39(d,j=7.2hz,2h),7.46(t,j=7.5hz,2h),7.51(t,j=7.5hz,1h),7.94(s,1h).
13
c nmr(151mhz,cdcl3):δ=13.9,60.9,117.6(q,j=267.6hz),126.5,128.1(2c),130.0,130.3(2c),133.2,134.0(q,j=2.5hz),135.9,161.6.
19
f nmr(470mhz,cdcl3):δ=-53.4(s,3f).hrms(esi-tof)calc’d for c
13h11
f3n2nao2[m+na]
+
:307.0665;found 307.0671。产物的氢谱图见图26,碳谱图见图27,氟谱图见图28;
[0102]
更换原料,可获得其他产物和产率如下图表:
[0103][0104]
实施例7:(n-cf3三氮唑的合成)
[0105]
氮气氛围下,向10ml schlenk封管中加入n-(n-三氟甲基亚胺酰基)吡啶盐(0.4mmol),重氮化合物(0.2mmol),dmf(2ml)和搅拌子,随后在0℃下滴加dbu的dmf溶液(90μl,0.6mmol溶于2ml的dmf中)反应30min后,恢复室温继续搅拌12h。加入二氯甲烷稀释后,用水洗涤(20ml
×
4),有机相用无水硫酸钠干燥,过滤,减压浓缩,硅胶柱层析分离提纯(石油醚/乙酸乙酯=30:1)得到n-cf3三氮唑类化合物;具体反应如下:
[0106][0107]
具体的,腈为苯甲腈,重氮化合物为重氮乙酸乙酯时,获得产物产率82%,其核磁和质谱表征:1h nmr(600mhz,cdcl3):δ=1.23(t,j=7.2hz,3h),4.30(q,j=7.2hz,2h),7.40(d,j=7.8hz,2h),7.52(t,j=7.5hz,2h),7.58(t,j=7.5hz,1h).
13
c nmr(151mhz,cdcl3):δ=13.9,61.6,117.7(q,j=271.4hz),123.5,128.4(2c),129.5(2c),130.9,138.3,141.0,159.5.
19
f nmr(470mhz,cdcl3):δ=-55.1(s,3f).hrms(esi-tof)calc’d for c
12h10
f3n3nao2[m+na]+:308.0617;found 308.0616.ir(in kbr,cm-1):1738,1429,1247,1207,1157。产物的氢谱图见图29,碳谱图见图30,氟谱图见图31;
[0108]
更换原料,可获得其他产物和产率如下:
[0109][0110]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。