1.本发明涉及金属氧氢化物的制造方法、金属氧氢化物、及使用其的氨合成方法。
2.本技术以于2019年7月8日在日本提出申请的特愿2019-127224号为基础来主张优先权,将其内容并入本文中。
背景技术:
3.在骨架中具有氢负离子(h-离子)的氧氢化物(oxyhydride)之中,也报道了显示出超导特性的lafeas(o
1-xhx
)、显示出氢负离子传导性的la
2-x-y
sr
x+y
lih
1-x+yo3-y
等,包含h-离子的物质受到瞩目。近年来,报道了将batio
3-xhx
、srtio
3-xhx
、catio
3-xhx
、lnho(ln=gd、sm)等氧氢化物与ru等金属纳米粒子组合而成的催化剂显示出高的氨合成活性(非专利文献1~3)。
4.另一方面,报道了baln2o4(ln=镧系元素(lanthanide))单晶的合成方法等(例如非专利文献4)。
5.现有技术文献
6.非专利文献
7.非专利文献1:kageyama等人.j.am.chem.soc.,2017,139,18240-18246页。
8.非专利文献2:kageyama等人。adv.energy mater 2018,1801772页。
9.非专利文献3:kageyama等人.j.am.chem.soc.,2018,140,11170-11173页。
10.非专利文献4:besara等人.progress in solid state chemistry(固体化学进展),2014,42,23-36页。
技术实现要素:
11.发明所要解决的课题
12.非专利文献1~3所公开的氧氢化物是由于源自h-离子的独特的特性而在各种领域受到关注的材料,但由于合成方法复杂,因此所合成的种类也还不多。在非专利文献4中,虽然有关于baln2o4的磁特性的记载,但并没有与将baln2o4的氧用氢负离子(h-离子)取代而得到的氧氢化物有关的公开,尤其没有公开使用该氧氢化物作为氨合成用催化剂。
13.用于解决课题的手段
14.本技术的发明人发现了在接近常压的反应条件下、以短时间使仅包含1种非氧元素的氧化物与金属氢化物反应从而制造金属氧氢化物的方法,从而完成了本发明。
15.即,本发明的要旨为以下内容。
16.〔1〕制造方法,其特征在于,其是在氢气氛下使氧化物与金属氢化物反应从而制造金属氧氢化物的方法,其中,
17.构成前述氧化物的非氧元素仅包含1种非氧元素,
18.前述反应的压力条件为0.1~0.9mpa,
19.前述反应的温度为500~1000℃。
20.〔2〕如〔1〕所述的制造方法,其中,前述金属氧氢化物包含:
21.构成前述金属氢化物的金属元素、
22.构成前述氧化物的前述非氧元素、
23.氧、和
24.氢,
25.前述金属元素与前述非氧元素为不同的元素。
26.〔3〕如〔1〕或〔2〕所述的制造方法,其包括下述工序:
27.混合工序,将前述氧化物与前述金属氢化物混合从而得到混合物;和
28.加热工序,在氢气氛下,以0.1~0.9mpa的压力和500~1000℃的温度对前述混合物进行加热。
29.〔4〕如〔3〕所述的制造方法,其还包括在前述混合工序之前对前述氧化物进行脱水处理的前处理工序。
30.〔5〕如〔1〕~〔4〕中任一项所述的制造方法,其中,前述氧化物为mmon(m为非氧元素,m为1或2;n表示2或3所示的数),mmon为选自由sc2o3、y2o3、lno2、zro2、tio2、sio2及al2o3组成的组中的1种,
31.前述金属氢化物为aeh2(ae:选自由mg、ca、ba、sr组成的组中的1种碱土金属),
32.前述金属氧氢化物为下述通式(1)所示的金属氧氢化物。
33.aerm
poq-xhy
ꢀꢀꢀ
(1)
34.(前述通式(1)中,ae为选自由mg、ca、ba、sr组成的组中的至少1种碱土金属;m为选自由sc、y、ln、zr、ti、si、及al组成的组中的1种非氧元素;r为1或2;p为1或2;q为3或4;x表示0.1≤x≤3.0所示的数;y表示0.2≤y≤3.0所示的数。)
35.〔6〕如〔5〕所述的制造方法,其中,前述金属氢化物为bah2,
36.前述通式(1)中,ae为ba。
37.〔7〕如〔5〕或〔6〕所述的制造方法,其中,前述氧化物与前述金属氢化物的装料摩尔比为p:0.5r~p:2.5r。
38.〔8〕下述通式(1)所示的金属氧氢化物。
39.aerm
poq-xhy
ꢀꢀꢀ
(1)
40.(前述通式(1)中,ae为选自由mg、ca、ba、sr组成的组中的至少1种碱土金属;m为选自由sc、y、ln、zr、ti、si、及al组成的组中的1种非氧元素;r为1或2;p为1或2;q为3或4;x表示0.1≤x≤3.0所示的数;y表示0.2≤y≤3.0所示的数。)
41.〔9〕如〔8〕所述的金属氧氢化物,其中,前述通式(1)中,ae为ba。
42.〔10〕如〔8〕或〔9〕所述的金属氧氢化物,其由下述通式(2)~(7)中的任一者表示。
43.aeln2o
4-xhy
ꢀꢀꢀ
(2)
44.ae2sio
4-xhy
ꢀꢀꢀ
(3)
45.aeal2o
4-xhy
ꢀꢀꢀ
(4)
46.aetio
3-xhy
ꢀꢀꢀ
(5)
47.aezro
3-xhy
ꢀꢀꢀ
(6)
48.(前述通式(2)~(6)中,ae、x、y表示与前述通式(1)中的ae、x、y相同的含义。)
49.〔11〕金属担载物,其特征在于,其是在担载体上担载过渡金属而得到的金属担载物,其中,
50.前述担载体为包含〔8〕~〔10〕中任一项所述的金属氧氢化物的组合物。
51.〔12〕如〔11〕所述的金属担载物,其中,前述过渡金属的担载量相对于前述担载体100质量份而言为0.01质量份以上且50质量份以下。
52.〔13〕如〔11〕或〔12〕所述的金属担载物,其中,前述过渡金属为选自由ru、co及fe组成的组中的至少一种。
53.〔14〕担载金属催化剂,其是由〔11〕~〔13〕中任一项所述的金属担载物形成的。
54.〔15〕氨合成用催化剂,其是由〔11〕~〔13〕中任一项所述的金属担载物形成的。
55.〔16〕氨合成用催化剂,其特征在于,其为包含〔8〕~〔10〕中任一项所述的金属氧氢化物的组合物。
56.〔17〕氨合成方法,其特征在于,在〔14〕所述的担载金属催化剂的存在下使氮与氢反应。
57.发明的效果
58.本发明的金属氧氢化物的制造方法能够在接近常压的反应条件下以短时间制造金属氧氢化物,因此作为金属氧氢化物的合成法是优选的。其与以往的金属氧氢化物的合成方法相比,能够在接近常压的反应条件下合成金属氧氢化物,从生产率及成本方面来看也优异。
59.另外,所得到的bace2o
4-xhy
等金属氧氢化物在用作氨合成用催化剂的情况下,即使在低反应温度且低反应压力下也具有高的氨合成活性,并且,即使反复进行合成反应,也不会观察到催化活性的降低。因此,该金属氧氢化物作为氨合成用催化剂是优选的。
附图说明
60.[图1]为示出本发明的一实施方式的制造金属氧氢化物的方法的图。
[0061]
[图2]为实施例1、4、5、6中得到的金属氧氢化物bace2o
4-xhy
粉末的xrd衍射图案。
[0062]
[图3]为实施例4中得到的金属氧氢化物bace2o
4-xhy
粉末的大气
·
水暴露前后xrd衍射图案。
[0063]
[图4]为示出实施例1、比较例4、5中的氨合成速度的反应温度依赖性的曲线图。
[0064]
[图5]为示出实施例2、3、7中的氨合成速度的反应温度依赖性的曲线图。
[0065]
[图6]为氢从实施例1、4中得到的金属氧氢化物bace2o
4-xhy
中的升温脱离谱图。
[0066]
[图7]为实施例8中得到的金属氧氢化物ba2sio
4-xhy
粉末的xrd衍射图案。
[0067]
[图8]为氢从实施例8中得到的金属氧氢化物ba2sio
4-xhy
中的升温脱离谱图。
[0068]
[图9]为实施例9中得到的金属氧氢化物baal2o
4-xhy
粉末的xrd衍射图案。
[0069]
[图10]为氢从实施例9中得到的金属氧氢化物baal2o
4-xhy
中的升温脱离谱图。
[0070]
[图11]为实施例10中得到的金属氧氢化物batio
3-xhy
粉末的xrd衍射图案。
[0071]
[图12]为氢从实施例10中得到的金属氧氢化物batio
3-xhy
中的升温脱离谱图。
[0072]
[图13]为实施例11中得到的金属氧氢化物bazro
3-xhy
粉末的xrd衍射图案。
[0073]
[图14]为氢从实施例11中得到的金属氧氢化物bazro
3-xhy
中的升温脱离谱图。
[0074]
[图15]为实施例12中得到的金属氧氢化物srzro
3-xhy
粉末的xrd衍射图案。
[0075]
[图16]为氢从实施例12中得到的金属氧氢化物srzro
3-xhy
中的升温脱离谱图。
具体实施方式
[0076]
以下,对本发明进行详细说明。
[0077]
(金属氧氢化物的制造方法)
[0078]
本发明的金属氧氢化物的制造方法是在氢气氛下使氧化物与金属氢化物反应从而制造金属氧氢化物的方法。构成前述氧化物的非氧元素仅包含1种非氧元素。前述反应的压力条件为0.1~0.9mpa,前述反应的温度为500~1000℃。
[0079]
优选地,通过本发明的制造方法得到的金属氧氢化物包含构成前述金属氢化物的金属元素、构成前述氧化物的前述非氧元素、氧、和氢,前述金属元素与前述非氧元素为不同的元素。
[0080]
另外,前述反应的压力条件优选为0.1~0.4mpa。前述反应的温度优选为600~800℃。前述反应时间优选为0.5~24小时。前述反应时间更优选为10~24小时。
[0081]
如图1所示,本发明的金属氧氢化物的制造方法的一实施方式优选包括下述工序:将前述氧化物与前述金属氢化物混合从而得到混合物的混合工序;以及,在氢气氛下,以0.1~0.9mpa的压力和500~1000℃的温度对前述混合物进行加热的工序。前述反应的压力条件为0.1~0.4mpa,前述反应的温度为600~800℃,前述反应时间优选为0.5~24小时。前述反应时间更优选为10~24小时。
[0082]
前述混合工序优选在例如氩等非活性气体气氛下进行。混合的方法没有特别限定,可举出使用玛瑙研钵进行混合的方法、使用球磨机进行混合的方法等。混合工序中使用的前述氧化物及前述金属氢化物优选为粉末状。例如,前述氧化物的平均粒径优选为数nm~数μm。前述金属氢化物的平均粒径优选为数十nm~数μm。
[0083]
使用的氧化物及金属氢化物的原料的方式优选为粉末。例如,在使用的氧化物的一个例子为ceo2的情况下,可举出市售的aldrich公司制的ceo2粉末(平均粒径:小于25nm)。在使用的金属氢化物的一个例子为bah2的情况下,可通过以下的步骤来合成。通过在氢气氛下对作为市售的试剂的ba金属(aridrich制,块状)进行加热等已知方法来进行合成,由此可得到bah2。
[0084]
前述混合工序中,对于氧化物与金属氢化物的装料比而言,例如在最终的金属氧氢化物由后述的下述通式(8)表示的情况下(即,r为1或2;p为1或2),优选非氧元素m与金属氢化物的金属x的摩尔比在p:0.5r(摩尔:摩尔)~p:2.5r(摩尔:摩尔)的范围内。例如,优选在p:0.7r(摩尔:摩尔)~p:1.1r(摩尔:摩尔)的范围内。可以是p:r(摩尔:摩尔),也可以是p:0.8r(摩尔:摩尔)。
[0085]
xrm
poq-xhy
ꢀꢀꢀ
(8)
[0086]
例如,前述氧化物和金属氢化物分别为ceo2和bah2的情况下,优选以ce与ba的摩尔比(ce:ba)成为0.5:1.0~1.0:0.5(mol/mol)的方式混合,更优选为0.8:1.0~1.0:0.8,进一步优选为0.95:1.0~1.0:0.95。即,关于合成的金属氧氢化物bace2o
4-xhy
,以使金属氢化物bah2的量过剩的方式添加。
[0087]
认为通过以使金属氢化物的量过剩的方式添加,从而混合物中的氢过剩地存在,即使在常压下,也能够以更短的时间进行合成反应。
[0088]
另一方面,例如,前述氧化物和金属氢化物分别为sio2和bah2的情况下,优选以si与ba的摩尔比(si:ba)成为1.0:1.5~1.0:2.5(mol/mol)的方式混合,更优选为1.0:1.8~1.0:2.2,进一步优选为1.0:1.95~1.0:2.05。即,关于合成的金属氧氢化物ba2sio
4-xhy
,以使金属氢化物bah2的量与金属氧氢化物中的si与ba的摩尔比(si:ba)相适应的方式添加。
[0089]
优选在混合工序之前还包括对作为要混合的原料的氧化物进行脱水处理的前处理工序。作为前述脱水处理工序,例如可举出在300℃以上且低于900℃、优选为400℃以上且低于800℃、更优选为500℃以上且低于700℃的温度下进行真空加热处理的方法。
[0090]
前述加热工序中,将通过前述混合工序得到的混合物配置在氢气氛中,于500~1000℃、优选600~800℃进行加热。加热工序的压力为接近常压的压力,即,为0.1~0.9mpa,更优选为0.1~0.7mpa,进一步优选为0.1~0.4mpa。
[0091]
加热方法没有特别限定,但优选在氢气流中加热。作为前述加热处理,例如可举出在氢气流中、于500~1000℃、优选600~800℃进行加热处理的方法。反应结束时间可以利用例如所得到的粉体的xrd衍射图案(x射线衍射图案)来判断。例如,反应时间优选为0.5~24小时,更优选为10~24小时。
[0092]
本发明的特征之一是无需以高压力进行加热处理。即使在接近大气压的压力条件下,也能够抑制释放出混合物中的氢,能够制造本发明的金属氧氢化物。
[0093]
关于其理由,尚未被阐明。预测为如下:在使1种过渡金属的氧化物与其他的金属氢化物进行金属氢化反应从而直接合成金属氧氢化物的情况下,与向过渡金属与其他金属的复合氧化物中导入氢从而间接合成金属氧氢化物的情况相比,所需要的能量变低。尤其认为,在其一实施方式中,以使金属氢化物的量过剩的方式添加的情况下,混合物中的氢过剩地存在,即使在常压下,也能够以更低的温度并且以更短的时间来进行合成反应。另外认为,作为原料而使用的氧化物及金属氢化物的粒径越小,则固相反应越进一步进行。
[0094]
[氧化物]
[0095]
对于本发明的制造方法涉及的氧化物而言,只要构成该氧化物的非氧元素为1种非氧元素,则没有特别限定。本发明的制造方法涉及的氧化物包括过渡金属氧化物和典型元素氧化物。
[0096]
本发明的制造方法涉及的氧化物优选为通式(9)所示的非氧元素m的氧化物。
[0097]mmon
ꢀꢀꢀ
(9)
[0098]
(前述通式(9)中,m表示选自由第一~第三过渡金属及第12族~第14族典型元素组成的组中的至少1种非氧元素;m为1或2;n为1~5。)
[0099]
作为构成本发明的制造方法涉及的氧化物的非氧元素m,可举出包括第一过渡金属、第二过渡金属和第三过渡金属的过渡金属、包括第12族典型元素、第13族典型元素和第14族典型元素的典型元素等。第一过渡金属的具体例包括sc(钪)、ti(钛)、v(钒)、cr(铬)、mn(锰)、fe(铁)、co(钴)、ni(镍)、及cu(铜)。第二过渡金属的具体例包括y(钇)、zr(锆)、nb(铌)、mo(钼)、tc(锝)、ru(钌)、rh(铑)、pd(钯)、及ag(银)。第三过渡金属的具体例包括ln(镧系元素)、hf(铪)、ta(钽)、w(钨)、re(铼)、os(锇)、ir(铱)、pt(铂)、及au(金)。第12族典型元素的具体例包括zn(锌)。第13族典型元素的具体例包括al(铝)、ga(镓)、in(铟)。第14族典型元素的具体例包括si(硅)、ge(锗)、sn(锡)。
[0100]
ln(镧系元素)为la(镧)、ce(铈)、pr(镨)、nd(钕)、pm(钷)、sm(钐)、eu(铕)、gd
(钆)、tb(铽)、dy(镝)、ho(钬)、er(铒)、tm(铥)、yb(镱)、或lu(镥)。
[0101]
构成本发明的制造方法涉及的氧化物的非氧元素m优选为sc(钪)、y(钇)、ln(镧系元素)、ti(钛)、zr(锆)、si(硅)、al(铝)。此处,作为前述的ln(镧系元素),更优选为la(镧)、ce(铈)、pr(镨)、sm(钐)。
[0102]
即,本发明的制造方法涉及的氧化物mmon优选为sc2o3、y2o3、lno2、tio2、zro2、sio2、al2o3。此处,作为前述的lno2,更优选为la2o3、ceo2、pro2、smo2。
[0103]
[金属氢化物]
[0104]
本发明的制造方法涉及的金属氢化物优选为通式(10)所示的金属元素x的氢化物(hydride)。
[0105]
xhnꢀꢀꢀ
(10)
[0106]
(前述通式(10)中,x表示选自元素周期表的第1族原子、第2族原子、第3族原子、或镧系元素原子中的至少1种,n表示1≤n≤3所示的数。)
[0107]
前述通式(10)中,x表示选自元素周期表的第1族原子、第2族原子、第3族原子、或镧系元素原子中的至少1种。
[0108]
用于前述x的原子没有特别限定,可以为1种,也可以包含2种以上的元素。包含2种以上的元素的情况下,没有特别限定,但优选包含同族的原子彼此、或镧系元素原子彼此。
[0109]
作为元素周期表的第2族原子(以下,简称为第2族原子,有时简记为ae。),没有特别限定,但优选为mg、ca、sr、ba,更优选为ba、ca、sr。进一步优选为ba。这是因为在将金属担载物用作后述的担载金属催化剂时的活性高。
[0110]
作为元素周期表的第3族原子(以下,称为第3族原子。),没有特别限定,但优选为y。这是因为其是存在量较多的元素。
[0111]
作为镧系元素原子,没有特别限定,但优选为la、ce、pr、nd、sm、eu、pr、yb。这是因为其为更为通用的材料。更优选为la、ce、nd、sm。这是因为其存在量较多。进一步优选为la、ce。这是因为在将金属担载物用作后述的担载金属催化剂时的活性高。
[0112]
x为镧系元素原子的情况下,可以包含多个镧系元素原子,具体而言,可以为稀土金属混合物(misch metal)。此处所谓“稀土金属混合物”,是含有多种稀土元素(稀土)的合金的通称,作为通常包含大量的ce作为其含有成分的合金而为人所知。
[0113]
需要说明的是,在下文中,有时将前述第3族原子和镧系元素原子总称并简记为re。
[0114]
作为前述x,优选为第2族原子、或镧系元素原子。这是因为其元素的存在量多,在将金属担载物用作后述的担载金属催化剂时的活性高。更优选为第2族原子。这是因为其元素的存在量多。
[0115]
另外,作为前述x,优选为ca、mg、sr、ba、y、或镧系元素原子,更优选为ca、mg、sr、ba、y、la、ce、pr、nd、sm、eu、pr、yb,进一步优选为ba、sr。
[0116]
前述通式(4)中的n表示1≤n≤3的数值。
[0117]
前述n在x为第1族原子的情况下没有特别限定,优选为1。前述n在x为第2族原子的情况下没有特别限定,优选为2。
[0118]
在x为第3族原子、或镧系元素原子的情况下,前述n通常表示2至3的任意数值,优选为2或3。
[0119]
前述ae及前述re通常形成离子键型氢化物。离子键型氢化物中包含的氢以氢负离子(h-离子)的形式存在,通过该氢与水、酸接触,从而生成氢(h2)和氢氧根离子(oh-)。
[0120]
就前述re的氢化物(以下,称为rehn)而言,作为通常的氢化物的二氢化物、和作为高密度氢化物的三氢化物这两者是已知的。而且,能够形成具有二氢化物与三氢化物之间的值的、高密度金属氢化物。需要说明的是,该高密度金属氢化物中,二氢化物与三氢化物之间的值也可连续变化。
[0121]
对于前述x而言,在不损害本发明的效果的情况下,其一部分可以还包含x以外的原子,具体而言,可以包含至少1种碱金属原子。
[0122]
本发明中可使用的金属氢化物没有特别限定,可以直接使用市售的试剂、工业原料,另外也可以使用通过将对应的金属在氢气氛下加热等已知方法进行合成而得到的产物。
[0123]
[金属氧氢化物]
[0124]
本发明的制造方法涉及的金属氧氢化物只要在氧化物的骨架中具有氢负离子(h-离子),则没有特别限定。例如,可以为下述通式(8)所示的金属氧氢化物。
[0125]
xrm
poq-xhy
ꢀꢀꢀ
(8)
[0126]
前述通式(8)中,x与上述金属氢化物的x同样;m与上述氧化物中的非氧元素同样;r为1或2;p为1或2;q为1~4的正数;x表示0.1≤x≤3.0所示的数;y表示0.2≤y≤3.0所示的数。x优选表示0.1≤x/r≤1.5所示的数;y优选表示0.1≤y/r≤1.5所示的数。对于h与x的摩尔比y/r而言,优选y/r≥0.1,对于h与x的摩尔比y/r而言,更优选y/r》0.5,进一步优选y/r≥1.0。
[0127]
作为本发明的金属氧氢化物涉及的一实施方式,可举出例如将由非氧元素(m)形成的复合氧化物的氧侧的一部分用氢负离子(h-离子)取代而形成的金属氧氢化物。其具体例为例如下述通式(1)所示的金属氧氢化物。
[0128]
aerm
poq-xhy
ꢀꢀꢀ
(1)
[0129]
前述通式(1)中,ae为选自由mg、ca、ba、及sr组成的组中的至少1种碱土金属;m为选自由sc、y、ln、zr、ti、si、及al组成的组中的1种非氧元素;r为1或2;p为1或2;q为正数3或4;x表示0.1≤x≤3.0所示的数;y表示0.2≤y≤3.0所示的数。x优选表示0.1≤x/r≤1.5所示的数;y优选表示0.1≤y/r≤1.5所示的数。对于h与ae的摩尔比y/r而言,优选y/r≥0.1,更优选y/r》0.5,进一步优选y/r≥1.0。
[0130]
前述通式(1)中,优选m为ln,r=1,p=2,q=4。例如,为下述通式(2)所示的金属氧氢化物。
[0131]
aeln2o
4-xhy
ꢀꢀꢀ
(2)
[0132]
前述通式(2)中,ae、x、y为与通式(1)相同的含义。
[0133]
另外,前述通式(2)中,优选地,ae为ba;ln为ce;x表示0.1≤x≤2.0所示的数;y表示0.2≤y≤2.0所示的数。即,为下述通式(11)所示的金属氧氢化物。更优选地,x表示0.2≤x≤1.5所示的数;y表示0.2≤y≤1.5所示的数。
[0134]
bace2o
4-xhy
ꢀꢀꢀ
(11)
[0135]
本发明的金属氧氢化物也可以是例如将由sio2、al2o3、tio2、zro2形成的复合氧化物的氧侧的一部分用氢负离子(h-离子)取代而形成的金属氧氢化物。其具体例分别包括以
下的通式(3)~(6)表示的金属氧氢化物。
[0136]
ae2sio
4-xhy
ꢀꢀꢀ
(3)
[0137]
aeal2o
4-xhy
ꢀꢀꢀ
(4)
[0138]
aetio
3-xhy
ꢀꢀꢀ
(5)
[0139]
aezro
3-xhy
ꢀꢀꢀ
(6)
[0140]
前述通式(3)~(6)中,ae、x、y为与通式(1)相同的含义。
[0141]
另外,前述通式(4)~(6)中,x优选表示0.1≤x≤2.0所示的数;y优选表示0.2≤y≤2.0所示的数。x更优选表示0.1≤x≤1.5所示的数;y更优选表示0.2≤y≤1.5所示的数。
[0142]
另外,本发明的金属氧氢化物也可以是例如将由氧化钒形成的复合氧化物的氧位点的一部分用氢负离子(h-离子)取代而形成的金属氧氢化物。其具体例为例如以下的通式(12)表示的金属氧氢化物。
[0143]
aevo
2-xhy
ꢀꢀꢀ
(12)
[0144]
通式(12)中,ae为例如ca、ba、sr等碱土金属。x表示0.1≤x≤1.5所示的数;y表示0.2≤y≤2.0所示的数。
[0145]
本发明的金属氧氢化物的具体例包括bala2o
4-xhy
、bapr2o
4-xhy
、basc2o
4-xhy
、basm2o
4-xhy
、bay2o
4-xhy
、casc2o
4-xhy
、mgsc2o
4-xhy
、srce2o
4-xhy
、srpr2o
4-xhy
、sry2o
4-xhy
、klao
2-xhy
、ksco
2-xhy
、kyo
2-xhy
、lilao
2-xhy
、liceo
2-xhy
、lisco
2-xhy
、liyo
2-xhy
、nasco
2-xhy
、nayo
2-xhy
、ba2sio
4-xhy
、baal2o
4-xhy
、batio
3-xhy
、bazro
3-xhy
、srzro
3-xhy
等。上述具体例中,x、y与通式(1)相同。
[0146]
本发明的金属氧氢化物中包含的氢负离子(h-离子)的量没有特别限定。金属氧氢化物为上述通式(8)所示的金属氧氢化物的情况下,优选金属氧氢化物的晶体结构维持不含氢负离子的下述通式(13)所示的复合氧化物的晶体结构。
[0147]
xrm
p
oqꢀꢀꢀ
(13)
[0148]
通式(13)中,x、m、r、p、q为与通式(8)相同的含义。
[0149]
例如,在为前述金属氧氢化物bace2o
4-xhy
的情况下,优选维持bace2o4的晶体结构。
[0150]
对于上述通式(8)所示的金属氧氢化物的x与y的关系而言,优选为x=(y/2)+σ以使金属氧氢化物为电荷中性。过渡金属m的价数恒定的情况下,优选σ=0。一部分过渡金属m的价数变化的情况下,为了维持电荷中性,σ例如表示-0.5≤σ≤+0.5所示的数。
[0151]
即,上述通式(8)、通式(1)、通式(2)~(6)、通式(11)及通式(12)也可以分别为以下的通式(8a)、通式(1a)、通式(2a)~(6a)、通式(11a)及通式(12a)。
[0152]
xrm
poq-(y/2)-σhy
ꢀꢀꢀ
(8a)
[0153]
aerm
poq-(y/2)-σhy
ꢀꢀꢀ
(1a)
[0154]
aeln2o
4-(y/2)-σhy
ꢀꢀꢀ
(2a)
[0155]
ae2sio
4-(y/2)-σhy
ꢀꢀꢀ
(3a)
[0156]
aeal2o
4-(y/2)-σhy
ꢀꢀꢀ
(4a)
[0157]
aetio
3-(y/2)-σhy
ꢀꢀꢀ
(5a)
[0158]
aezro
3-(y/2)-σhy
ꢀꢀꢀ
(6a)
[0159]
bace2o
4-(y/2)-σhy
ꢀꢀꢀ
(11a)
[0160]
aevo
2-(y/2)-σhy
ꢀꢀꢀ
(12a)
[0161]
上述各式中,x、m、ae、r、p、q、y的含义与通式(8)、通式(1)、通式(2)~(6)、通式(11)及通式(12)相同,σ表示-0.5≤σ≤+0.5所示的数。σ优选表示-0.25≤σ≤+0.25所示的数。
[0162]
例如,后述的实施例1中,合成了bace2o
3.33h1.34
(600℃)及bace2o
3.62h0.76
(800℃)(其中,使全部ce的氧化值为3价。)所示的金属氧氢化物。在认为ce的氧化值变化的情况下,可以将这些金属氧氢化物分别用bace2o
3.33-σh1.34
及bace2o
3.62-σh0.76
表示(σ的含义与上述通式相同)。
[0163]
后述的实施例8中,合成了ba2sio2h
2.68e1.32
所示的金属氧氢化物(其中,认为氧的值最大为2,假设在阴离子位点存在电子的情况)。在认为si的氧化值变化的情况下,可以将这些金属氧氢化物用ba2sio
2.66-δh2.68
表示(σ的含义与上述通式相同)。
[0164]
后述的实施例9中,合成了baal2o3h
0.22e1.78
所示的金属氧氢化物(其中,认为氧的值最大为3,假设在阴离子位点存在电子的情况)。在认为al的氧化值变化的情况下,可以将这些金属氧氢化物用baal2o
3.89-δh0.22
表示(σ的含义与上述通式相同)。
[0165]
后述的实施例10中,合成了batio2h
1.33
所示的金属氧氢化物(其中,认为氧的值最大为2,假设在阴离子位点存在电子的情况)。在认为ti的氧化值变化的情况下,可以将这些金属氧氢化物用batio
2.33-δh1.33
表示(σ的含义与上述通式相同)。
[0166]
后述的实施例11中,合成了bazro2h
1.62e0.38
所示的金属氧氢化物(其中,认为氧的值最大为2,假设在阴离子位点存在电子的情况)。在认为zr的氧化值变化的情况下,可以将这些金属氧氢化物用bazro
2.19-δh1.62
表示(σ的含义与上述通式相同)。
[0167]
后述的实施例12中,合成了srzro2h
1.86e0.13
所示的金属氧氢化物(其中,认为氧的值最大为2,假设在阴离子位点存在电子的情况)。在认为zr的氧化值变化的情况下,可以将这些金属氧氢化物用srzro
2.07-δh1.86
表示(σ的含义与上述通式相同)。
[0168]
《金属氧氢化物中包含的氢负离子(h-离子)的定量》
[0169]
可以利用升温脱离分析装置(belcata)对所合成的金属氧氢化物进行分析,由此求出脱离的氢量。基于脱离的氢量的结果,可得到金属氧氢化物中包含的氢负离子(h-离子)的比例。例如,在后文的实施例中,基于利用升温脱离分析装置(belcata)进行分析而得到的结果(图6),可以将于600℃合成的bace2o
4-xhy
表示为bace2o
3.33h1.34
(其中,使全部ce的氧化值为3价。),另外,可以将于800℃合成的bace2o
4-xhy
表示为bace2o
3.62h0.76
(其中,使全部ce的氧化值为3价。)。
[0170]
(金属担载物)
[0171]
本发明的金属担载物是在担载体上担载过渡金属而得到的产物。
[0172]
前述担载体为包含上述的本发明的金属氧氢化物的组合物。本发明的一实施方式的金属担载物(以下,有时也称为本实施方式的金属担载物。)是在担载体上担载过渡金属而得到的产物。前述担载体优选为下述通式(1)所示的金属氧氢化物。
[0173]
aerm
poq-xhy
ꢀꢀꢀ
(1)
[0174]
前述通式(1)中,ae为选自由mg、ca、ba、sr组成的组中的至少1种碱土金属;m为选自由sc、y、ln、zr、ti、si、及al组成的组中的1种非氧元素;r为1或2;p为1或2;q为正数3或4;x表示0.1≤x≤3.0所示的数;y表示0.2≤y≤3.0所示的数。x优选表示0.1≤x/r≤1.5所示的数;y优选表示0.1≤y/r≤1.5所示的数。对于h与ae的摩尔比y/r而言,优选y/r≥0.1,更
优选y/r》0.5,进一步优选y/r≥1.0。
[0175]
前述通式(1)中,优选m为ln,r=1,p=2,q=4。前述担载体例如优选为下述通式(2)所示的金属氧氢化物。
[0176]
aeln2o
4-xhy
ꢀꢀꢀ
(2)
[0177]
前述通式(2)中,ae、x、y为与通式(1)相同的含义。
[0178]
另外,前述通式(2)中,优选地,ae为ba;ln为ce;x表示0.1≤x≤2.0所示的数;y表示0.2≤y≤2.0所示的数。即,为下述通式(11)所示的金属氧氢化物。更优选地,x表示0.2≤x≤1.5所示的数;y表示0.2≤y≤1.5所示的数。
[0179]
bace2o
4-xhy
ꢀꢀꢀ
(11)
[0180]
前述担载体也可以是例如将由sio2、al2o3、tio2、zro2形成的复合氧化物的氧侧的一部分取代为氢负离子(h-离子)而形成的金属氧氢化物。其具体例分别包括以下的通式(3)~(6)表示的金属氧氢化物。
[0181]
ae2sio
4-xhy
ꢀꢀꢀ
(3)
[0182]
aeal2o
4-xhy
ꢀꢀꢀ
(4)
[0183]
aetio
3-xhy
ꢀꢀꢀ
(5)
[0184]
aezro
3-xhy
ꢀꢀꢀ
(6)
[0185]
前述通式(3)~(6)中,ae、x、y为与通式(1)相同的含义。
[0186]
另外,前述通式(4)~(6)中,x优选表示0.1≤x≤2.0所示的数;y优选表示0.2≤y≤2.0所示的数。x更优选表示0.1≤x≤1.5所示的数;y更优选表示0.2≤y≤1.5所示的数。
[0187]
本发明的金属担载物涉及的前述过渡金属的担载量没有特别限定,优选相对于前述担载体100质量份而言为0.01质量份以上且50质量份以下。
[0188]
另外,本发明的金属担载物涉及的前述过渡金属的担载量没有特别限定,但相对于该催化剂的总量而言,通常为0.5wt%以上,优选为1wt%以上,更优选为2wt%以上,通常为30wt%以下,优选为20wt%以下,更优选为10wt%以下。若为前述下限值以上,则可得到本发明的效果,若为前述上限值以下,则可得到担载量与成本均衡的本发明的效果。
[0189]
本实施方式的金属担载物是在担载体上担载过渡金属而得到的产物。前述担载体优选为包含通过在氢气氛下对上述氧化物和上述金属氢化物进行加热而得到的上述金属氧氢化物的组合物。更优选为包含通过下述制造方法得到的金属氧氢化物的组合物,所述制造方法包括下述工序:混合工序,将上述氧化物与上述金属氢化物混合;和加热处理工序,在氢气氛中对通过混合工序得到的混合物进行加热处理。优选还包括在前述混合工序之前进一步对上述氧化物进行脱水处理的前处理工序。在该情况下,对于前述混合工序而言,优选在ar手套箱中等稀有气体气氛中将经脱水处理的氧化物与金属氢化物混合。此外,优选的加热处理温度、优选的加热时间、优选的原料的装入量比等与上述本发明的金属氧氢化物的制造方法相同。
[0190]
《过渡金属》
[0191]
本发明的金属担载物涉及的过渡金属没有特别限定,通常为元素周期表的第6族、第7族、第8族、第9族、第10族的过渡金属,优选为第6族、第8族、或第9族的过渡金属,更优选为第8族或第9族金属。
[0192]
另外,作为具体的金属元素,没有特别限定,通常为cr、mo、mn、re、fe、ru、os、co、
rh、ni、pd、pt。优选为mo、re、fe、ru、os、co。这是因为这些金属元素与氮的键能高。更优选为ru、co或fe。这是因为在将氨合成用催化剂作为氨合成用催化剂来使用时,具有氨合成活性。进一步优选为ru。这是因为其具有最高的催化活性。
[0193]
前述的各金属元素可以单独使用,也可以组合2种以上而使用。另外,也可以使用这些金属元素的金属间化合物、例如co3mo3n、fe3mo3n、ni2mo3n、mo2n等。优选为各金属元素单独或2种以上的组合,更优选单独使用,这在成本方面是有利的。
[0194]
《过渡金属向金属氧氢化物上的担载方法》
[0195]
过渡金属向金属氧氢化物上的担载方法没有特别限定,例如,可以将由前述方法得到的粉末状金属氧氢化物(例如,bace2o
4-xhy
)、和要担载的金属的化合物填入二氧化硅玻璃管内,将其在氢+氮气流中(n2:h2=1:3,流量:8ml/min)升温2小时直至200℃。其后继续升温2小时直至400℃,接着于400℃加热2小时,由此,得到在粉末状金属氧氢化物上固定有过渡金属me(例如,ru)的担载物(以下为me/金属氧氢化物,例如ru/bace2o
4-xhy
)。
[0196]
例如,通过使用过渡金属分别为ru、co、fe的过渡金属化合物ru3(co)
12
、co2(co)
8、
fe2(co)9,能够合成金属担载物即担载有ru的bace2o
4-xhy
(缩写为ru/bace2o
4-xhy
)、担载有co的bace2o
4-xhy
(缩写为co/bace2o
4-xhy
)、担载有fe的bace2o
4-xhy
(缩写为fe/bace2o
4-xhy
)、担载有ru的ba2sio
4-xhy
(缩写为ru/ba2sio
4-xhy
)、担载有ru的baal2o
4-xhy
(缩写为ru/baal2o
4-xhy
)、担载有ru的batio
3-xhy
(缩写为ru/tio
3-xhy
)、担载有ru的bazro
3-xhy
(缩写为ru/bazro
3-xhy
)、担载有ru的srzro
3-xhy
(缩写为ru/srzro
3-xhy
)等。
[0197]
《金属担载物的形状》
[0198]
本实施方式的金属担载物的形状没有特别限定,具体而言,可以为块状、粉末状、被膜状等任意形状,通常为粉末状。粉末状的金属担载物的粒径没有特别限定,通常为1nm以上且10μm以下。
[0199]
本实施方式的金属担载物中的过渡金属的粒径没有特别限定,通常为1nm以上且100nm以下。优选为20nm以下,更优选为10nm以下。这是因为,在作为氨合成用催化剂来使用时,作为氮解离的活性部位的阶跃位点(step site)数变多。
[0200]
(担载金属催化剂)
[0201]
本实施方式的担载金属催化剂是由上述金属担载物形成的。本实施方式的担载金属催化剂包含过渡金属和对前述过渡金属进行担载的担载体,前述担载体为前述的金属氧氢化物。例如,前述担载体优选为下述通式(1)所示的金属氧氢化物。
[0202]
aerm
poq-xhy
ꢀꢀꢀ
(1)
[0203]
前述通式(1)中,ae为选自由mg、ca、ba、sr组成的组中的至少1种碱土金属;m为选自由sc、y、ln、zr、ti、si、及al组成的组中的1种非氧元素;r为1或2;p为1或2;q为正数3或4;x表示0.1≤x≤3.0所示的数;y表示0.2≤y≤3.0所示的数。x优选表示0.1≤x/r≤1.5所示的数;y优选表示0.1≤y/r≤1.5所示的数。对于h与ae的摩尔比y/r而言,优选y/r≥0.1,更优选y/r》0.5,进一步优选y/r≥1.0。
[0204]
(氨合成用催化剂)
[0205]
本发明的氨合成用催化剂是在担载体上担载过渡金属而得到的产物。本实施方式的氨合成用催化剂包含过渡金属和对前述过渡金属进行担载的担载体。前述担载体优选为包含前文说明的金属氧氢化物的组合物。例如,前述担载体优选为包含下述通式(1)所示的
金属氧氢化物的组合物。
[0206]
aerm
poq-xhy
ꢀꢀꢀ
(1)
[0207]
前述通式(1)中,ae为选自由mg、ca、ba、sr组成的组中的至少1种碱土金属;m为选自由sc、y、ln、zr、ti、si、及al组成的组中的1种非氧元素;r为1或2;p为1或2;q为正数3或4;x表示0.1≤x≤3.0所示的数;y表示0.2≤y≤3.0所示的数。x优选表示0.1≤x/r≤1.5所示的数;y优选表示0.1≤y/r≤1.5所示的数。对于h与ae的摩尔比y/r而言,优选y/r≥0.1,更优选y/r》0.5,进一步优选y/r≥1.0。
[0208]
《过渡金属》
[0209]
本实施方式中使用的过渡金属没有特别限定,通常为元素周期表的第6族、第7族、第8族、第9族、第10族的过渡金属,优选为第6族、第8族、或第9族的过渡金属,更优选为第8族或第9族金属。
[0210]
另外,作为具体的金属元素,没有特别限定,通常为cr、mo、mn、re、fe、ru、os、co、rh、ni、pd、pt。优选为mo、re、fe、ru、os、co。这是因为这些金属元素与氮的键能高。更优选为ru、co或fe。这是因为在将氨合成用催化剂作为氨合成用催化剂来使用时,具有氨合成活性。进一步优选为ru。这是因为其具有最高的催化活性。
[0211]
前述的各金属元素可以单独使用,也可以组合2种以上而使用。另外,也可以使用这些金属元素的金属间化合物、例如co3mo3n、fe3mo3n、ni2mo3n、mo2n等。优选为各金属元素单独或2种以上的组合,更优选单独使用,这在成本方面是有利的。
[0212]
(氨合成用催化剂的制造方法)
[0213]
本发明的氨合成用催化剂是在担载体上担载过渡金属而得到的产物。前述担载体为包含前述的本发明的金属氧氢化物的组合物。本实施方式的氨合成用催化剂通过使前述过渡金属担载于包含含有前述金属氧氢化物的组合物的前述担载体而制造。制造方法没有特别限定,通常,向前述担载体上担载过渡金属、或作为过渡金属的前体的化合物(以下为过渡金属化合物)而制造。
[0214]
成为本实施方式的氨合成用催化剂的原料的、前述金属氧氢化物的组合物可以直接使用市售的试剂、工业原料,另外,也可以使用通过已知方法由对应的金属合成得到的产物。
[0215]
可以在对本实施方式中使用的前述金属氧氢化物组合物实施在氢气氛中于200~500℃左右加热数小时、例如于340℃加热2小时的前处理之后,通过后述的过渡金属担载工序使其担载前述过渡金属。
[0216]
对于使用将前述担载体预先在氢气氛下加热而得到的试样制造的催化剂而言,例如在用于氨合成反应的情况下,在反应开始后可立即得到高活性。
[0217]
使过渡金属担载于本实施方式中可使用的前述担载体的方法没有特别限定,可以使用已知方法。通常使用下述方法:使作为要担载的过渡金属的化合物的、能够通过还原、热分解等而转化为过渡金属的过渡金属化合物担载于前述担载体后转化为过渡金属。
[0218]
作为前述过渡金属化合物,没有特别限定,可以使用容易发生热分解的过渡金属的无机化合物或有机过渡金属配合物等。具体而言,可以使用过渡金属的配合物、过渡金属的氧化物、硝酸盐、盐酸盐等过渡金属盐等。
[0219]
例如,作为ru化合物,可举出十二羰基三钌[ru3(co)
12
]、二氯四(三苯基膦)钌(ii)
[rucl2(pph3)4]、二氯三(三苯基膦)钌(ii)[rucl2(pph3)3]、三(乙酰丙酮)钌(iii)[ru(acac)3]、二茂钌[ru(c5h5)]、亚硝酰基硝酸钌[ru(no)(no3)3]、钌酸钾、氧化钌、硝酸钌、氯化钌等。优选为三(乙酰丙酮)钌(iii)[ru(acac)3]。
[0220]
作为fe化合物,可举出五羰基铁[fe(co)5]、十二羰基三铁[fe3(co)
12
]、九羰基铁[fe2(co)9]、四羰基碘化铁[fe(co)4i2]、三(乙酰丙酮)铁(iii)[fe(acac)3]、二茂铁[fe(c5h5)2]、氧化铁、硝酸铁、氯化铁(fecl3)等。
[0221]
作为co化合物,可举出八羰基钴[co2(co)8]、三(乙酰丙酮)钴(iii)[co(acac)3]、乙酰丙酮钴(ii)[co(acac)2]、二茂钴[co(c5h5)2]、氧化钴、硝酸钴、氯化钴等。
[0222]
这些过渡金属化合物中,对于[ru3(co)
12
]、[fe(co)5]、[fe3(co)
12
]、[fe2(co)9]、[co2(co)8]等过渡金属的羰基配合物而言,通过在担载之后进行加热,得以担载过渡金属,因此,在制造本实施方式的氨合成用催化剂时,可以省略后述的还原处理,从这一点考虑是优选的。
[0223]
前述过渡金属化合物的使用量没有特别限定,可以适当地使用用于实现所期望的担载量的量。通常,相对于所使用的前述担载体的质量而言,通常为2wt%以上,优选为10wt%以上,更优选为20wt%以上,通常为50wt%以下,优选为40wt%以下,更优选为30wt%以下。
[0224]
作为使前述过渡金属化合物担载于担载体的方法,具体而言,可举出例如物理性的混合法、cvd法(化学沉积法)、溅射法等方法。
[0225]
物理性的混合法为下述方法:在将前述担载体、与前述过渡金属化合物以固体形态混合后,在氮、氩、氦等非活性气体气流中、或在真空下进行加热。此时的加热温度没有特别限定,通常为200℃以上且600℃以下。加热时间没有特别限定,通常优选为2小时以上。
[0226]
此处,若为可通过热分解而转化为过渡金属的过渡金属化合物,则在该阶段通常可担载过渡金属,所得到的担载物成为本实施方式的氨合成用催化剂。
[0227]
在使用除了可通过热分解而转化为过渡金属的过渡金属化合物以外的化合物的情况下,通常对过渡金属化合物进行还原,由此,所得到的还原处理物成为本实施方式的氨合成用催化剂。
[0228]
对前述过渡金属化合物进行还原的方法(以下,称为还原处理)只要不妨碍本发明的目的,则没有特别限定,可举出例如:在包含还原性气体的气氛下进行的方法;在包含前述过渡金属化合物的溶液中加入nabh4、nh2nh2或、福尔马林等还原剂,使其在前述金属氢化物的表面析出的方法。优选为在包含还原性气体的气氛下进行的方法。作为前述还原性气体,可举出氢、氨、甲醇(蒸气)、乙醇(蒸气)、甲烷、乙烷等。
[0229]
另外,在前述还原处理时,也可以在反应体系中共存有不妨碍本发明的目的、特别是不妨碍氨合成反应的、除还原性气体以外的成分。具体而言,在还原处理时,除了氢等还原性气体以外还可以共存有不妨碍反应的氩、氮等气体。优选使氮共存。
[0230]
在包含氢的气体中进行前述还原处理的情况下,通过与氢一同存在氮,能够与后述的氨的制造并行地进行该还原处理。即,在将本实施方式的氨合成用催化剂用作后述的氨合成用催化剂的情况下,可以通过将使前述过渡金属化合物担载于前述金属氢化物而得到的产物置于氨合成反应的反应条件中,从而对前述过渡金属化合物进行还原而使其转化为过渡金属。
[0231]
前述还原处理时的温度没有特别限定,通常为200℃以上,优选为300℃以上。另外,更优选可于低于700℃的温度进行。进一步优选为400℃以上且低于700℃。这是因为,通过在前述的还原处理温度范围内进行,前述过渡金属的生长充分并且在优选的范围内进行。
[0232]
前述还原处理时的压力没有特别限定,通常为0.01mpa以上且10mpa以下。对于还原处理时的压力而言,若设定为与后述的氨合成条件相同的条件,则不需要繁琐的操作,在制造效率方面是有利的。
[0233]
前述还原处理的时间没有特别限定,在于常压下实施的情况下,通常为1小时以上,优选为2小时以上。
[0234]
另外,在反应压力高的条件、例如在1mpa以上的条件下进行的情况下,作为前述还原处理的时间,优选为1小时以上。
[0235]
在使用除了可通过热分解而转化为过渡金属的过渡金属化合物以外的化合物的情况下,可以与前述的还原处理方法同样地,利用通常的方法对固体混合物中包含的过渡金属化合物进行还原,由此得到本实施方式的氨合成用催化剂。
[0236]
作为除前述金属氧氢化物及前述过渡金属以外的成分,也可以进一步包含sio2、al2o3、zro2、mgo、活性炭、石墨、sic等作为前述金属氧氢化物的担载体。
[0237]
本实施方式的氨合成用催化剂可以以使用通常的成型技术进行成型而得到的成型体的形状使用。具体而言,可举出粒状、球状、片、环、通心粉、四叶、骰子、蜂窝状等形状。另外,本实施方式的氨合成用催化剂也可以涂覆于适当的支承体后使用。
[0238]
使用本实施方式的氨合成用催化剂时,其反应活性没有特别限定,以反应温度为300℃、反应压力为0.9mpa时的氨的生成速度为例的情况下,优选为1.0mmol/g
·
h以上。更优选为2.0mmol/g
·
h以上。这是因为其适合于实用的制造条件。进一步优选为3.0mmol/g
·
h以上。这是因为其适合于更高效的制造条件。最优选为5.0mmol/g
·
h以上。这是因为其适合于进一步高效的制造条件。
[0239]
以下,对使用本实施方式的氨合成用催化剂的氨的制造方法进行说明。
[0240]
(氨的制造方法(氨合成方法))
[0241]
本实施方式的氨的制造方法(以下,有时称为本实施方式的制造方法)是在本实施方式的担载金属催化剂存在下使氮与氢反应的方法。即,为下述方法:使用本实施方式的担载金属催化剂或本实施方式的氨合成用催化剂作为催化剂,使氢与氮在前述催化剂上反应而合成氨。
[0242]
作为具体的制造方法,只要是使氢与氮在前述催化剂上接触而合成氨的方法,则没有特别限定,可以适当地依照已知的制造方法来制造。
[0243]
本实施方式的氨的制造方法中,通常,在使氢与氮在前述催化剂上接触时,对催化剂进行加热,由此制造氨。
[0244]
本实施方式的制造方法中的反应温度没有特别限定,通常为200℃以上,优选为250℃以上,更优选为300℃以上,通常为600℃以下,优选为500℃以下,更优选为450℃以下。氨合成是放热反应,因此,低温区域的反应在化学平衡理论上对氨的生成更有利,但为了得到充分的氨生成速度,优选在上述的温度范围内进行反应。
[0245]
本实施方式的制造方法中,与前述催化剂接触的氮与氢的摩尔比率没有特别限
定,通常,作为氢相对于氮的比率(h2/n2(体积/体积)),通常为0.4以上,优选为0.5以上,更优选为1以上,通常为10以下,优选为5以下。
[0246]
本实施方式的制造方法中的反应压力没有特别限定,作为包含氮和氢的混合气体的压力,通常为0.01mpa以上,优选为0.1mpa以上,通常为20mpa以下,优选为15mpa以下,更优选为10mpa以下。另外,若考虑实用性的利用,优选为大气压以上的加压条件。
[0247]
本实施方式的制造方法中,优选的是,在使氮和氢与前述催化剂接触之前,将附着于前述催化剂的水分、氧化物通过使用脱水材料的方法、进行深冷分离的方法或使用氢气等除去。作为除去的方法,可举出还原处理。
[0248]
本实施方式的制造方法中,为了得到更良好的氨收率,优选本实施方式的制造方法中使用的氮及氢中的水分含量少。本实施方式的制造方法中使用的氮及氢中的水分含量没有特别限定,通常优选的是,氮与氢的混合气体中的总水分含量为100ppm以下,优选为50ppm以下。
[0249]
本实施方式的制造方法中,反应容器的形式没有特别限定,可以使用通常可用于氨合成反应的反应容器。作为具体的反应形式,可以使用例如批处理式反应形式、封闭循环系反应形式、流通系反应形式等。其中,从实用性的观点考虑,优选为流通系反应形式。另外,也可以使用将填充有催化剂的一种反应器、或多种反应器连结的方法、在同一反应器内具有多种反应层的反应器的任意方法。
[0250]
由氢和氮合成氨的反应是伴有体积收缩的放热反应,因此,为了提高氨收率,在工业上优选除去反应热,可以使用带有通常所用的除热机构的已知的反应装置。例如,具体而言,可以使用下述方法等:将填充有催化剂的反应器以串联的方式连结多个,在各反应器的出口处设置中间冷却器来进行除热。
[0251]
(氨合成用催化剂的其他实施方式)
[0252]
[由金属氧氢化物粉末形成的氨合成用催化剂]
[0253]
也可以将前述实施方式中得到的金属氧氢化物粉末在不担载过渡金属的情况下直接作为氨合成用催化剂来使用(不含担载金属)。前述金属氧氢化物粉末优选为包含下述通式(2)所示的金属氧氢化物的组合物。
[0254]
aeln2o
4-xhy
ꢀꢀꢀ
(2)
[0255]
前述通式(2)中,ae为选自由mg、ca、ba、sr组成的组中的至少1种碱土金属;x表示0.1≤x≤3.0所示的数;y表示0.2≤y≤3.0所示的数。x优选表示0.1≤x≤1.5所示的数;y优选表示0.1≤y≤1.5所示的数。对于y而言,优选y≥0.1,更优选y》0.5,进一步优选y≥1.0。
[0256]
另外,前述金属氧氢化物粉末更优选为包含下述通式(11)所示的金属氧氢化物的组合物。
[0257]
bace2o
4-xhy
ꢀꢀꢀ
(11)
[0258]
前述通式(11)中,x、y为与通式(1)相同的含义。另外,优选地,x表示0.2≤x≤1.5所示的数;y表示0.2≤y≤1.5所示的数。
[0259]
[使用金属氧氢化物粉末的氨合成]
[0260]
《氨合成反应》
[0261]
可在与前述实施方式相同的条件下实施氨合成反应。例如,如使用bace2o
4-xhy
粉末的后述的实施例7及图5所示,400℃、0.9mpa时的氨的合成速度为0.4mmol/g/hr。500℃、
0.9mpa时的氨的合成速度为1.7mmol/g/hr。
[0262]
本实施方式的氨的制造方法中,可以单独使用通过本实施方式的制造方法得到的氨合成用催化剂,也可以与通常可用于氨合成的其他的已知催化剂组合而使用。
[0263]
实施例
[0264]
以下,基于实施例,对本发明进行更详细的说明。使用气相色谱仪对nh3的生成量进行定量,或者,使生成的nh3溶解于硫酸水溶液中后,将所得的溶液作为试样,使用离子色谱仪对该试样中包含的nh3的量进行定量,由此,求出氨生成速度,基于所得到的氨生成速度来进行氨合成活性的评价。
[0265]
(离子色谱分析)
[0266]
使用离子色谱仪,对通过使从反应容器排出的氨气溶解于5mm硫酸水溶液而捕获的铵离子(nh
4+
)进行分析。分析条件如下所述。
[0267]
[测定条件]
[0268]
装置:岛津制作所公司制prominence
[0269]
检测器:电导率检测器cdd-10avp(岛津制作所公司制)
[0270]
柱:离子色谱用柱ic-c4(岛津制作所公司制)
[0271]
洗脱液:3.0mm草酸+2.0mm 18-冠-6-醚水溶液流速:1.0ml/分钟
[0272]
柱温:40℃
[0273]
(实施例1)
[0274]
(氨合成用催化剂的制备)
[0275]
《金属氧氢化物bace2o
4-xhy
粉末的合成》
[0276]
于600℃对ceo2进行真空加热处理,由此将吸附于表面的水等除去。在ar手套箱中,使用玛瑙研钵,将所得到的脱水处理后的ceo2、与bah2以ce与ba的摩尔比成为1:1的方式混合。在h2气流中,于600℃对所得到的混合物的粉体进行20小时加热处理,由此得到红褐色的粉末状的bace2o
4-xhy
。
[0277]
[ru向bace2o
4-xhy
上的担载]
[0278]
将由前述方法得到的粉末状的bace2o
4-xhy 0.50g、和ru3(co)
12
(aldrich公司制,99%)0.056g(相对于bace2o
4-xhy
而言,以要担载的金属ru计相当于5质量%)填入二氧化硅玻璃管内,将其在氢+氮气流中(n2:h2=1:3,流量:8ml/min)升温2小时直至200℃。其后继续升温2小时直至400℃,接着于400℃加热2小时,由此得到在bace2o
4-xhy
上固定有ru的担载物(以下为ru/bace2o
4-xhy
)。
[0279]
接下来,在以下的实施例中,使用所得到的担载物作为氨合成用催化剂,进行氨合成。
[0280]
[使用担载有ru的bace2o
4-xhy
的氨合成]
[0281]
《氨合成反应》
[0282]
将前述ru/bace2o
4-xhy
作为催化剂,使该催化剂接触氮与氢的混合气体,由此进行氨合成反应。将前述ru/bace2o
4-xhy 0.1g装在sus制反应管中,使用具备其的固定床流通式反应装置进行氨合成反应。原料的氮气的水分浓度和原料的氢气的水分浓度各自为检测限以下。关于该反应时的两种原料气体的流量,氮为15ml/min,氢为45ml/min(共计60ml/min)。另外,该反应时的反应压力为0.9mpa,反应温度为300℃,反应时间为30小时。
[0283]
《氨的生成速度》
[0284]
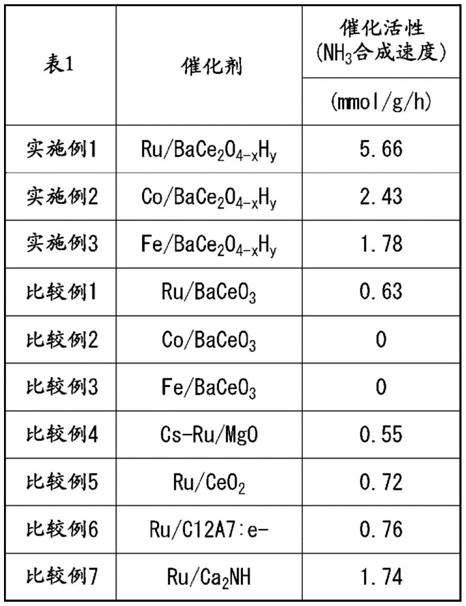
使从前述固定床流通式反应装置出来的气体在0.005m硫酸水溶液中鼓泡,由此使前述气体中的氨溶解于前述硫酸水溶液中,然后,通过使用离子色谱仪的前述方法对产生的铵离子进行定量。使用离子色谱仪,经时性地对由氨合成反应生成的氨的生成速度进行了测定,结果,氨生成速度为5.66mmol/g/hr。该值为远远高于后述的比较例1中得到的ru/baceo3(0.63mmol/g/hr)的值。将结果示于表1。
[0285]
另外,改变前述氨合成反应的反应温度来进行同样的实验,由此对氨生成速度的反应温度依赖性进行评价。将结果示于图4。
[0286]
(实施例2)
[0287]
[co向bace2o
4-xhy
上的担载]
[0288]
将由前述方法得到的粉末状的bace2o
4-xhy 95mg、和co2(co)
8 14.5mg(相对于bace2o
4-xhy
而言,以要担载的金属co计相当于5质量%)装入石英玻璃反应管中,在向其中流通氮15ml/min和氢45ml/min(共计60ml/min)的同时,升温2小时直至400℃。接下来,于该温度维持5小时,由此得到在bace2o
4-xhy
上固定有co的担载物(以下为co/bace2o
4-xhy
)。
[0289]
接下来,在以下的实施例中,使用所得到的担载物作为氨合成用催化剂,进行氨合成。
[0290]
[使用担载有co的bace2o
4-xhy
的氨合成]
[0291]
《氨合成反应》
[0292]
代替实施例1中使用的ru/bace2o
4-xhy
,而使用前述co/bace2o
4-xhy
作为催化剂,除此以外,利用与实施例1同样的方法及条件,进行生成氨(nh3)的反应(以下为氨合成反应)。
[0293]
《氨的生成速度》
[0294]
利用与实施例1同样的方法,使用离子色谱仪,经时性地对由氨合成反应生成的氨的生成速度进行了测定,结果,氨生成速度为2.43mmol/g/hr。将结果示于表1。
[0295]
另外,利用与实施例1同样的方法,对氨生成速度的反应温度依赖性进行评价。将结果示于图5。
[0296]
(实施例3)
[0297]
[fe向bace2o
4-xhy
上的担载]
[0298]
将由前述方法得到的粉末状的bace2o
4-xhy 95mg、和fe2(co)
9 16.3mg(相对于bace2o
4-xhy
而言,以要担载的金属fe计相当于5质量%)装入石英玻璃反应管中,在向其中流通氮15ml/min和氢45ml/min(共计60ml/min)的同时,升温2小时直至400℃。接下来,于该温度维持5小时,由此得到在bace2o
4-xhy
上固定有fe的担载物(以下为fe/bace2o
4-xhy
)。
[0299]
接下来,在以下的实施例中,使用所得到的担载物作为氨合成用催化剂,进行氨合成。
[0300]
[使用担载有fe的bace2o
4-xhy
的氨合成]
[0301]
《氨合成反应》
[0302]
代替实施例1中使用的ru/bace2o
4-xhy
,而使用前述fe/bace2o
4-xhy
作为催化剂,除此以外,利用与实施例1同样的方法及条件,进行生成氨(nh3)的反应(以下为氨合成反应)。
[0303]
《氨的生成速度》
[0304]
利用与实施例1同样的方法,使用离子色谱仪,经时性地对由氨合成反应生成的氨
的生成速度进行了测定,结果,氨生成速度为1.78mmol/g/hr。将结果示于表1。
[0305]
另外,利用与实施例1同样的方法,对氨生成速度的反应温度依赖性进行评价。将结果示于图5。
[0306]
(比较例1)
[0307]
[baceo3粉末的合成]
[0308]
使硝酸钡5.23g(0.02mol)和六水合硝酸铈8.68g(0.02mol)及柠檬酸38.4g(0.2mol)溶解于水中。向所得到的水溶液中加入二甘醇42.4g(0.4mol)后,搅拌一小时。其后,于120℃对所得到的混合物进行4小时加热,使其凝胶化。其后,于450℃对所得到的凝胶化物进行5小时加热,使其碳化。于900℃对已碳化的混合粉体进行6小时加热,由此制备粉末状的baceo3。
[0309]
[ru向baceo3上的担载]
[0310]
使用与实施例1同样的方法,以金属ru相对于baceo3而言成为5质量%的方式进行担载,由此制备担载物ru/baceo3。
[0311]
接下来,在以下的实施例中,使用所得到的担载物作为氨合成用催化剂,进行氨合成。
[0312]
《氨合成反应》
[0313]
代替实施例1中使用的ru/bace2o
4-xhy
,而使用前述的ru/baceo3作为催化剂,除此以外,利用与实施例1同样的方法及条件,进行氨合成反应。
[0314]
《氨的生成速度》
[0315]
利用与实施例1同样的方法,使用离子色谱仪,经时性地对由氨合成反应生成的氨的生成速度进行了测定,结果,氨的生成速度为0.63mmol/g/hr。将结果示于表1。
[0316]
(比较例2)
[0317]
[co向baceo3上的担载]
[0318]
使用与比较例1同样的方法,以金属co相对于baceo3而言成为5质量%的方式进行担载,由此制备担载物co/baceo3。
[0319]
[使用co/baceo3的氨合成]
[0320]
《氨合成反应》
[0321]
使用前述的co/baceo3作为催化剂,除此以外,利用与实施例1同样的方法及条件,进行氨合成反应。
[0322]
《氨的生成速度》
[0323]
利用与实施例1同样的方法,使用离子色谱仪,经时性地对由氨合成反应生成的氨的生成速度进行了测定,结果,氨的生成速度为0mmol/g/hr。将结果示于表1。
[0324]
(比较例3)
[0325]
[fe向baceo3上的担载]
[0326]
使用与比较例1同样的方法,以金属fe相对于baceo3而言成为5质量%的方式进行担载,由此制备担载物fe/baceo3。
[0327]
[使用fe/baceo3的氨合成]
[0328]
《氨合成反应》
[0329]
使用前述的fe/baceo3作为催化剂,除此以外,利用与实施例1同样的方法及相同
条件,进行氨合成反应。
[0330]
《氨的生成速度》
[0331]
利用与实施例1同样的方法,使用离子色谱仪,经时性地对由氨合成反应生成的氨的生成速度进行了测定,结果,氨的生成速度为0mmol/g/hr。将结果示于表1。
[0332]
(比较例4)
[0333]
[ru向cs/mgo上的担载]
[0334]
代替实施例1中使用的bace2o
4-xhy
,而使用添加有cs的mgo(表述为cs/mgo),除此以外,利用与实施例1同样的方法,制备5wt%cs-ru/mgo催化剂(cs/ru元素比=1)。
[0335]
[使用cs-ru/mgo的氨合成]
[0336]
《氨合成反应》
[0337]
利用与实施例1同样的方法及条件,实施氨合成反应。300℃、0.9mpa时的氨的合成速度如表1所示,为0.55mmol/g/hr。将结果示于表1。
[0338]
另外,利用与实施例1同样的方法及条件,对氨生成速度的反应温度依赖性进行评价。将结果示于图4。
[0339]
(比较例5)
[0340]
[ru向ceo2上的担载]
[0341]
代替实施例1中使用的bace2o
4-xhy
而使用ceo2,除此以外,利用与实施例1同样的方法,制备5wt%ru/ceo2催化剂。
[0342]
[使用ru/ceo2的氨合成]
[0343]
《氨合成反应》
[0344]
利用与实施例1同样的方法及条件,实施氨合成反应。300℃、0.9mpa时的氨的合成速度如表1所示,为0.72mmol/g/hr。将结果示于表1。
[0345]
另外,利用与实施例1同样的方法及条件,对氨生成速度的反应温度依赖性进行评价。将结果示于图4。
[0346]
(比较例6)
[0347]
[ru向c12a7:e-上的担载]
[0348]
代替实施例1中使用的bace2o
4-xhy
而使用c12a7:e-,除此以外,利用与实施例1同样的方法,制备2wt%ru/c12a7:e-催化剂。
[0349]
[使用ru/c12a7:e-的氨合成]
[0350]
《氨合成反应》
[0351]
利用与实施例1同样的方法及条件,实施氨合成反应。300℃、0.9mpa时的氨的合成速度如表1所示,为0.76mmol/g/hr。将结果示于表1。
[0352]
(比较例7)
[0353]
[ru向ca2n上的担载]
[0354]
代替实施例1中使用的bace2o
4-xhy
而使用ca2n,除此以外,利用与实施例1同样的方法,制备5wt%ru/ca2n催化剂。
[0355]
[使用ru/ca2n的氨的合成]
[0356]
《氨合成反应》
[0357]
利用与实施例1同样的方法及条件,实施氨合成反应。300℃、0.9mpa时的氨的合成
速度如表1所示,为1.74mmol/g/hr。将结果示于表1。
[0358]
(实施例4~6)
[0359]
[于各种温度合成的bace2o
4-xhy
粉末的评价]
[0360]
代替实施例1中设定的氢气流中的加热处理温度即600℃,而分别采用表2所示的加热处理温度,除此以外,利用与实施例1同样的方法,制备bace2o
4-xhy
粉末。
[0361]
《bace2o
4-xhy
粉末的xrd》
[0362]
将利用上述方法而在实施例1、4~6中合成的试样的xrd衍射图案示于图2。在于比600℃低的温度合成的试样中,观察到多个峰混合存在的衍射图案,未得到单相的试样。另一方面,在于600℃以上合成的试样中,由观察到的衍射图案可知,得到了与bace2o4大致相同的单相的材料。另外可知,利用本方法合成的试样与bace2o4相比,峰均向低角度侧位移。认为这是因为,离子半径大的氢进入了bace2o4的氧的位点。此外可知,即使将实施例4中合成的bace2o
4-xhy
粉末在大气中或水中保存2天,xrd衍射图案也完全不变,处于试样的颜色(红褐色)也得以维持的状态(图3)。而且确认到,本材料是在水、大气中也稳定的氧氢化物。
[0363]
《bace2o
4-xhy
中包含的氢的定量》
[0364]
将利用升温脱离分析装置(belcata)来分析于600℃(实施例1)及800℃(实施例4)合成的bace2o
4-xhy
而得到的结果示于图6。从100℃附近起观察到氢的脱离,在700℃附近显示最大值,直到800℃左右为止观察到氢的脱离。根据脱离的氢量,于600℃及800℃合成的试样分别可以表示为bace2o
3.33h1.34
、bace2o
3.62h0.76
(其中,使全部ce的氧化值为3价。)。(由于使全部ce的氧化值为3价,因此认为氧的值最大为3。另外,若考虑到电荷平衡而假设在阴离子位点存在电子的情况下,也可以表示为bace2o3h
1.34e1.67
、bace2o3h
0.76e1.38
。推测有下述两种可能性:该电子孤立存在,或者使ce、ba的价数减少。)
[0365]
[ru向bace2o
4-xhy
上的担载]
[0366]
代替实施例1中使用的bace2o
4-xhy
,而使用实施例4~6中得到的粉末状的bace2o
4-xhy
,除此以外,利用与实施例1同样的方法,得到在bace2o
4-xhy
上固定有ru的担载物(以下为ru/bace2o
4-xhy
)。
[0367]
[使用ru/bace2o
4-xhy
的氨合成]
[0368]
《氨合成反应》
[0369]
将实施例4中使用的ru/bace2o
4-xhy
用作催化剂,除此以外,利用与实施例1同样的方法及条件,进行氨合成反应。补充说明的是,在实施例5和实施例6这两者中,所使用的bace2o
4-xhy
中包含大量杂质,因此没有对其催化活性进行调查。
[0370]
《氨的生成速度》
[0371]
利用与实施例1同样的方法,使用离子色谱仪,经时性地对由氨合成反应生成的氨的生成速度进行了测定,结果,氨的合成速度为1.15mmol/g/hr。实施例4中的氨生成速度比实施例1中的氨生成速度慢。
[0372]
(实施例7)
[0373]
[由bace2o
4-xhy
粉末形成的氨合成用催化剂]
[0374]
将实施例1中得到的bace2o
4-xhy
的粉末在不担载过渡金属的情况下直接作为氨合成用催化剂来使用(不含担载金属)。
[0375]
[使用bace2o
4-xhy
粉末的氨合成]
[0376]
《氨合成反应》
[0377]
除了采用400℃以外,利用与实施例1同样的方法及条件,进行氨合成反应。400℃、0.9mpa时的氨的合成速度为0.4mmol/g/hr。另外,利用与实施例1同样的方法及条件,对氨生成速度的反应温度依赖性进行评价。将结果示于图5。
[0378]
[表1]
[0379][0380]
表1的实施例及比较例的反应条件如下。
[0381]
催化剂量:0.1g,反应温度:300℃,反应气体流量:60ml/min,反应气体组成:n2/h2=1/3(v/v),反应压力:0.9mpa。
[0382]
[表2]
[0383][0384]
(实施例8)
[0385]
《ba2sio
4-xhy
粉末的合成》
[0386]
于600℃对sio2进行真空加热处理,由此将吸附于表面的水等除去。在ar手套箱中,使用玛瑙研钵,将所得到的脱水处理后的sio2、与bah2以si与ba的摩尔比成为1:2的方式
混合。在h2气流中,于650℃对所得到的混合物的粉体进行20小时加热处理,由此得到黑色的粉末状的ba2sio
4-xhy
。
[0387]
《ba2sio
4-xhy
粉末的xrd》
[0388]
将利用上述方法合成的试样的xrd衍射图案示于图7。在该试样中,由观察到的衍射图案可知,得到了与ba2sio4大致相同的单相的材料。另外可知,利用本方法合成的试样与ba2sio4相比,峰均向低角度侧位移。认为这是因为,形成了价数低于si
4+
的si,离子半径变大。
[0389]
《ba2sio
4-xhy
中包含的氢的定量》
[0390]
将利用升温脱离分析装置(belcata)对所合成的ba2sio
4-xhy
进行分析而得到的结果示于图8。从50℃附近起观察到氢的脱离,在730℃附近显示最大值,直到800℃左右为止观察到氢的脱离。根据脱离的氢的量,所合成的试样可以表示为ba2sio
2.66h2.68
(其中,使全部si的氧化值为4价。)。(由于原料为sio2,因此认为氧的值最大为2。另外,若考虑到电荷平衡而假设在阴离子位点存在电子的情况下,也可以表示为ba2sio2h
2.68e1.32
。推测有下述两种可能性:该电子孤立存在,或者使si、ba的价数减少。)
[0391]
[ru向ba2sio
4-xhy
上的担载]
[0392]
将由前述方法得到的粉末状的ba2sio
4-xhy 0.50g、和ru3(co)
12
(aldrich公司制,99%)0.056g(相对于ba2sio
4-xhy
而言,以要担载的金属ru计相当于5质量%)填入二氧化硅玻璃管内,将其在氢+氮气流中(n2:h2=1:3,流量:8ml/min)升温2小时直至200℃。其后继续升温2小时直至400℃,接着于400℃加热2小时,由此得到在ba2sio
4-xhy
上固定有ru的担载物(以下为ru/ba2sio
4-xhy
)。
[0393]
接下来,在以下的实施例中,使用所得到的担载物作为氨合成用催化剂,进行氨合成。
[0394]
[使用担载有ru的ba2sio
4-xhy
的氨合成]
[0395]
《氨合成反应》
[0396]
将前述ru/ba2sio
4-xhy
作为催化剂,使该催化剂接触氮与氢的混合气体,由此进行氨合成反应。将前述ru/ba2sio
4-xhy 0.1g装在sus制反应管中,使用具备其的固定床流通式反应装置进行氨合成反应。原料的氮气的水分浓度和原料的氢气的水分浓度各自为检测限以下。关于该反应时的两种原料气体的流量,氮为15ml/min,氢为45ml/min(共计60ml/min)。另外,该反应时的反应压力为0.9mpa,反应温度为300℃,反应时间为30小时。
[0397]
《氨的生成速度》
[0398]
使从前述固定床流通式反应装置出来的气体在0.005m硫酸水溶液中鼓泡,由此使前述气体中的氨溶解于前述硫酸水溶液中,然后,通过使用离子色谱仪的前述方法对产生的铵离子进行定量。使用离子色谱仪,经时性地对由氨合成反应生成的氨的生成速度进行了测定,结果,氨生成速度为10.95mmol/g/hr。将结果示于表3。
[0399]
[表3]
[0400][0401]
表3的实施例的反应条件如下。
[0402]
催化剂量:0.1g,反应温度:300℃,反应气体流量:60ml/min,
[0403]
反应气体组成:n2/h2=1/3(v/v),反应压力:0.9mpa。
[0404]
(实施例9)
[0405]
《baal2o
4-xhy
粉末的合成》
[0406]
于600℃对al2o3进行真空加热处理,由此将吸附于表面的水等除去。在ar手套箱中,使用玛瑙研钵,将所得到的脱水处理后的al2o3与bah2以al与ba的摩尔比成为2:1的方式混合。在h2气流中,于800℃对所得到的混合物的粉体进行20小时加热处理,由此得到黑色的粉末状的baal2o
4-xhy
。
[0407]
《baal2o
4-xhy
粉末的xrd》
[0408]
将利用上述方法合成的试样的xrd衍射图案示于图9。在该试样中,由观察到的衍射图案可知,得到了与baal2o4大致相同的单相的材料。另外可知,利用本方法合成的试样与baal2o4相比,峰均向低角度侧位移。认为这是因为,形成了价数低于al
3+
的al,离子半径变大。
[0409]
《baal2o
4-xhy
中包含的氢的定量》
[0410]
将利用升温脱离分析装置(belcata)对所合成的baal2o
4-xhy
进行分析而得到的结果示于图10。从50℃附近起观察到氢的脱离,在600℃附近显示最大值,直到1000℃左右为止观察到氢的脱离。根据脱离的氢的量,所合成的试样可以表示为baal2o
3.89h0.22
(其中,使全部al的氧化值为3价。)。(由于原料为al2o3,因此认为氧的值最大为3。另外,若考虑到电荷平衡而假设在阴离子位点存在电子的情况下,也可以表示为baal2o3h
0.22e1.78
。推测有下述两种可能性:该电子孤立存在,或者使al、ba的价数减少。)
[0411]
[ru向baal2o
4-xhy
上的担载]
[0412]
将由前述方法得到的粉末状的baal2o
4-xhy 0.50g、和ru3(co)
12
(aldrich公司制,99%)0.056g(相对于baal2o
4-xhy
而言,以要担载的金属ru计相当于5质量%)填入二氧化硅玻璃管内,将其在氢+氮气流中(n2:h2=1:3,流量:8ml/min)升温2小时直至200℃。其后继续升温2小时直至400℃,接着于400℃加热2小时,由此得到在baal2o
4-xhy
上固定有ru的担载物(以下为ru/baal2o
4-xhy
)。
[0413]
接下来,在以下的实施例中,使用所得到的担载物作为氨合成用催化剂,进行氨合成。
[0414]
[使用担载有ru的baal2o
4-xhy
的氨合成]
[0415]
《氨合成反应》
[0416]
将前述ru/baal2o
4-xhy
作为催化剂,使该催化剂接触氮与氢的混合气体,由此进行氨合成反应。将前述ru/baal2o
4-xhy 0.1g装在sus制反应管中,使用具备其的固定床流通式反应装置进行氨合成反应。原料的氮气的水分浓度和原料的氢气的水分浓度各自为检测限以下。关于该反应时的两种原料气体的流量,氮为15ml/min,氢为45ml/min(共计60ml/min)。另外,该反应时的反应压力为0.9mpa,反应温度为300℃,反应时间为30小时。
[0417]
《氨的生成速度》
[0418]
使从前述固定床流通式反应装置出来的气体在0.005m硫酸水溶液中鼓泡,由此使前述气体中的氨溶解于前述硫酸水溶液中,然后,通过使用离子色谱仪的前述方法对产生的铵离子进行定量。使用离子色谱仪,经时性地对由氨合成反应生成的氨的生成速度进行了测定,结果,氨生成速度为2.20mmol/g/hr。将结果示于表3。
[0419]
(实施例10)
[0420]
《batio
3-xhy
粉末的合成》
[0421]
于600℃对tio2进行真空加热处理,由此将吸附于表面的水等除去。在ar手套箱中,使用玛瑙研钵,将所得到的脱水处理后的tio2、与bah2以ti与ba的摩尔比成为1:1的方式混合。在h2气流中,于800℃对所得到的混合物的粉体进行20小时加热处理,由此得到黑色的粉末状的batio
3-xhy
。
[0422]
《batio
3-xhy
粉末的xrd》
[0423]
将利用上述方法合成的试样的xrd衍射图案示于图11。在该试样中,由观察到的衍射图案可知,得到了与batio3大致相同的单相的材料。另外可知,利用本方法合成的试样与batio3相比,峰均向低角度侧位移。认为这是因为,形成了价数低于ti
4+
的ti,离子半径变大。
[0424]
《batio
3-xhy
中包含的氢的定量》
[0425]
将利用升温脱离分析装置(belcata)对所合成的batio
3-xhy
进行分析而得到的结果示于图12。从150℃附近起观察到氢的脱离,在500℃附近显示最大值,直到950℃左右为止观察到氢的脱离。根据脱离的氢的量,所合成的试样可以表示为batio
2.33h1.33
(其中,使全部ti的氧化值为4价。)。由此可知,较之阴山组(非专利文献1~2)所报道的物质(batio
2.5h0.5
)而言以更高的浓度导入了氢负离子。(由于原料为tio2,因此认为氧的值最大为2。另外,若考虑到电荷平衡而假设在阴离子位点存在电子的情况下,也可以表示为batio2h
1.33e0.67
。推测有下述两种可能性:该电子孤立存在,或者使ti、ba的价数减少。)
[0426]
[ru向batio
3-xhy
上的担载]
[0427]
将由前述方法得到的粉末状的batio
3-xhy 0.50g、和ru3(co)
12
(aldrich公司制,99%)0.056g(相对于batio
3-xhy
而言,以要担载的金属ru计相当于5质量%)填入二氧化硅玻璃管内,将其在氢+氮气流中(n2:h2=1:3,流量:8ml/min)升温2小时直至200℃。其后继续升温2小时直至400℃,接着于400℃加热2小时,由此得到在batio
3-xhy
上固定有ru的担载物(以下为ru/batio
3-xhy
)。
[0428]
接下来,在以下的实施例中,使用所得到的担载物作为氨合成用催化剂,进行氨合成。
[0429]
[使用担载有ru的batio
3-xhy
的氨合成]
[0430]
《氨合成反应》
[0431]
将前述ru/batio
3-xhy
作为催化剂,使该催化剂接触氮与氢的混合气体,由此进行氨合成反应。将前述ru/batio
3-xhy 0.1g装在sus制反应管中,使用具备其的固定床流通式反应装置进行氨合成反应。原料的氮气的水分浓度和原料的氢气的水分浓度各自为检测限以下。关于该反应时的两种原料气体的流量,氮为15ml/min,氢为45ml/min(共计60ml/min)。另外,该反应时的反应压力为0.9mpa,反应温度为300℃,反应时间为30小时。
[0432]
《氨的生成速度》
[0433]
使从前述固定床流通式反应装置出来的气体在0.005m硫酸水溶液中鼓泡,由此使前述气体中的氨溶解于前述硫酸水溶液中,然后,通过使用离子色谱仪的前述方法对产生的铵离子进行定量。使用离子色谱仪,经时性地对由氨合成反应生成的氨的生成速度进行了测定,结果,氨生成速度为3.67mmol/g/hr。将结果示于表3。
[0434]
(实施例11)
[0435]
《bazro
3-xhy
粉末的合成》
[0436]
于600℃对zro2进行真空加热处理,由此将吸附于表面的水等除去。在ar手套箱中,使用玛瑙研钵,将所得到的脱水处理后的zro2、与bah2以zr与ba的摩尔比成为1:1的方式混合。在h2气流中,于800℃对所得到的混合物的粉体进行20小时加热处理,由此得到黑色的粉末状的bazro
3-xhy
。
[0437]
《bazro
3-xhy
粉末的xrd》
[0438]
将利用上述方法合成的试样的xrd衍射图案示于图13。在该试样中,由观察到的衍射图案可知,得到了与bazro3大致相同的单相的材料。然而,可知作为杂质而形成了zrh2。另外可知,利用本方法合成的试样与bazro3相比,峰均略微向高角度侧位移。其理由不甚明了。
[0439]
《bazro
3-xhy
中包含的氢的定量》
[0440]
将利用升温脱离分析装置(belcata)对所合成的bazro
3-xhy
进行分析而得到的结果示于图12。从50℃附近起观察到氢的脱离,在600℃附近显示最大值,直到1000℃左右为止观察到氢的脱离。根据脱离的氢的量,所合成的试样可以表示为bazro
2.19h1.62
(其中,使全部zr的氧化值为4价。)。(由于原料为zro2,因此认为氧的值最大为2。另外,若考虑到电荷平衡而假设在阴离子位点存在电子的情况下,也可以表示为bazro2h
1.62e0.38
。推测有下述两种可能性:该电子孤立存在,或者使zr、ba的价数减少。)
[0441]
[ru向bazro
3-xhy
上的担载]
[0442]
将由前述方法得到的粉末状的bazro
3-xhy 0.50g、和ru3(co)
12
(aldrich公司制,99%)0.056g(相对于bazro
3-xhy
而言,以要担载的金属ru计相当于5质量%)填入二氧化硅玻璃管内,将其在氢+氮气流中(n2:h2=1:3,流量:8ml/min)升温2小时直至200℃。其后继续升温2小时直至400℃,接着于400℃加热2小时,由此得到在bazro
3-xhy
上固定有ru的担载物(以下为ru/bazro
3-xhy
)。
[0443]
接下来,在以下的实施例中,使用所得到的担载物作为氨合成用催化剂,进行氨合成。
[0444]
[使用担载有ru的bazro
3-xhy
的氨合成]
[0445]
《氨合成反应》
[0446]
将前述ru/bazro
3-xhy
作为催化剂,使该催化剂接触氮与氢的混合气体,由此进行氨合成反应。将前述ru/bazro
3-xhy 0.1g装在sus制反应管中,使用具备其的固定床流通式反应装置进行氨合成反应。原料的氮气的水分浓度和原料的氢气的水分浓度各自为检测限以下。关于该反应时的两种原料气体的流量,氮为15ml/min,氢为45ml/min(共计60ml/min)。另外,该反应时的反应压力为0.9mpa,反应温度为300℃,反应时间为30小时。
[0447]
《氨的生成速度》
[0448]
使从前述固定床流通式反应装置出来的气体在0.005m硫酸水溶液中鼓泡,由此使前述气体中的氨溶解于前述硫酸水溶液中,然后,通过使用离子色谱仪的前述方法对产生的铵离子进行定量。使用离子色谱仪,经时性地对由氨合成反应生成的氨的生成速度进行了测定,结果,氨生成速度为3.73mmol/g/hr。将结果示于表3。
[0449]
(实施例12)
[0450]
《srzro
3-xhy
粉末的合成》
[0451]
于600℃对zro2进行真空加热处理,由此将吸附于表面的水等除去。在ar手套箱中,使用玛瑙研钵,将所得到的脱水处理后的zro2、与srh2以zr与sr的摩尔比成为1:1的方式混合。在h2气流中,于800℃对所得到的混合物的粉体进行20小时加热处理,由此得到黑色的粉末状的srzro
3-xhy
。
[0452]
《srzro
3-xhy
粉末的xrd》
[0453]
将利用上述方法合成的试样的xrd衍射图案示于图15。在该试样中,由观察到的衍射图案可知,得到了与srzro3大致相同的单相的材料。然而,可知作为杂质而形成了zrh2。另外可知,利用本方法合成的试样与srzro3相比,峰均略微向高角度侧位移。其理由不甚明了。
[0454]
《srzro
3-xhy
中包含的氢的定量》
[0455]
将利用升温脱离分析装置(belcata)对所合成的bazro
3-xhy
进行分析而得到的结果示于图16(数据获取中)。从50℃附近起观察到氢的脱离,在700~800℃附近显示最大值,直到1000℃左右为止观察到氢的脱离。根据脱离的氢的量,所合成的试样可以表示为srzro
2.07h1.86
(其中,使全部zr的氧化值为4价。)。(由于原料为zro2,因此认为氧的值最大为2。另外,若考虑到电荷平衡而假设在阴离子位点存在电子的情况下,也可以表示为srzro2h
1.86e0.13
。推测有下述两种可能性:该电子孤立存在,或者使zr、ba的价数减少。)
[0456]
[ru向srzro
3-xhy
上的担载]
[0457]
将由前述方法得到的粉末状的srzro
3-xhy 0.50g、和ru3(co)
12
(aldrich公司制,99%)0.056g(相对于srzro
3-xhy
而言,以要担载的金属ru计相当于5质量%)填入二氧化硅玻璃管内,将其在氢+氮气流中(n2:h2=1:3,流量:8ml/min)升温2小时直至200℃。其后继续升温2小时直至400℃,接着于400℃加热2小时,由此得到在srzro
3-xhy
上固定有ru的担载物(以下为ru/bazro
3-xhy
)。
[0458]
接下来,在以下的实施例中,使用所得到的担载物作为氨合成用催化剂,进行氨合成。
[0459]
[使用担载有ru的srzro
3-xhy
的氨合成]
[0460]
《氨合成反应》
[0461]
将前述ru/srzro
3-xhy
作为催化剂,使该催化剂接触氮与氢的混合气体,由此进行氨合成反应。将前述ru/srzro
3-xhy 0.1g装在sus制反应管中,使用具备其的固定床流通式反应装置进行氨合成反应。原料的氮气的水分浓度和原料的氢气的水分浓度各自为检测限以下。关于该反应时的两种原料气体的流量,氮为15ml/min,氢为45ml/min(共计60ml/min)。另外,该反应时的反应压力为0.9mpa,反应温度为300℃,反应时间为30小时。
[0462]
《氨的生成速度》
[0463]
使从前述固定床流通式反应装置出来的气体在0.005m硫酸水溶液中鼓泡,由此使前述气体中的氨溶解于前述硫酸水溶液中,然后,通过使用离子色谱仪的前述方法对产生的铵离子进行定量。使用离子色谱仪,经时性地对由氨合成反应生成的氨的生成速度进行了测定,结果,氨生成速度为3.56mmol/g/hr。将结果示于表3。