1.本发明属于有机合成领域,特别涉及一种2-芳基苯乙酮类化合物的的合成方法。
背景技术:
2.2-芳基苯乙酮类化合物是一类重要的医药和香料中间体,在药物化学和有机合成等领域有着广泛的应用。例如,已上市的三苯氧胺、大豆苷元和奥卡西平都含有2-芳基苯乙酮基。尽管2-芳基苯乙酮类化合物已经有多种合成方法,但仍需要更有效的合成方法。
3.当前合成2-芳基苯乙酮类化合物的合成方法主要有以下几种:
4.第一种是需要提前制备有机金属试剂(rli,rmgbr),再与韦伯酰胺可以形成稳定的四面体中间体,防止了酮羰基的形成和亲核试剂的过度加成。具体路线如下
[0005][0006]
第二种是以卤代芳烃原料,经过渡金属催化与苯乙酮进行α-芳基化反应生成目标产物(hamann,b.c.;hartwig,j.f.palladium-catalyzeddirectα-arylationofketones.rateaccelerationbystericallyhinderedchelatingligandsandreductiveeliminationfromatransitionmetalenolatecomplex.j.am.chem.soc.1997,119,12382-12383)
[0007][0008]
对于以上两种方法分析可知,这两种方法要么需要提前制备金属试剂(格式试剂,锂试剂),要么是需要使用过渡金属催化剂。因此,我们需要开发一种无过度金属,在温和条件下,一锅法合成2-芳基苯乙酮类化合物的方法。
技术实现要素:
[0009]
本发明的目的在于提供一种合成方法简单、反应条件温和、经济、绿色环保、适用性广泛或适于规模化生产2-芳基苯乙酮类化合物。
[0010]
实现上述目的的技术方案如下:
[0011]
一种2-芳基苯乙酮类化合物的合成方法,式1所示的n-甲氧基-n-甲基苯甲酰胺类、苯甲酸甲酯类化合物和式2所示的甲苯类化合物,布朗斯特碱,铯盐添加剂,有机溶剂,反应选择性合成式3所示的2-芳基苯乙酮类化合物。
[0012][0013]
其中,生成2-芳基苯乙酮类化合物中的r1选自氢、卤素基;r2选自氢、卤素基。
[0014]
优选的,r1为氢,r2为氢或卤素基。
[0015]
更加优选的,r1为氢,r2为氢。
[0016]
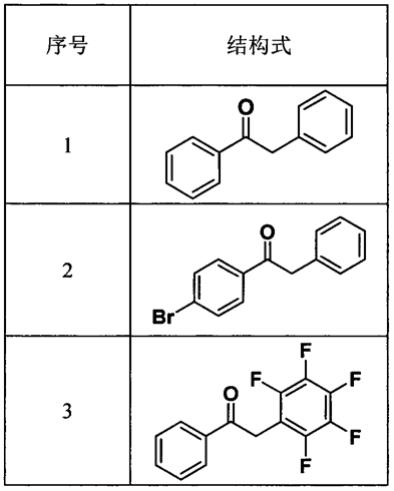
优选的,合成的2-芳基苯乙酮类化合物为如下结构中的一种。
[0017][0018]
优选的,生成2-芳基苯乙酮类化合物的反应中,式1所示的苯甲酸甲酯类化合物、铯盐添加剂、布朗斯特碱的摩尔比为:1∶(1~3)∶(2~5),式1所示的n-甲氧基-n-甲基苯甲酰胺类化合物、铯盐添加剂、布朗斯特碱的摩尔比为:1∶(1~3)∶(2~5),式1所示的苯甲酸甲酯类化合物、n-甲氧基-n-甲基苯甲酰胺类化合物与式2所示的甲苯类化合物摩尔比为:1∶(50~60)。
[0019]
优选的,布朗斯特碱为双(三甲基硅基)氨基钠、双(三甲基硅基氨基锂)、双(三甲基硅烷基)氨基钾。
[0020]
更加优选的,布朗斯特碱为双(三甲基硅基)氨基锂。
[0021]
优选的,所述的反应在有机溶剂中,以难以进行化学反应的气体保护进行反应。
[0022]
优选的,所述的难以进行化学反应的气体为氩气或氮气;有机溶剂为异丙醚、环戊基甲基醚、四氢呋喃、2-甲基四氢呋喃、甲基叔丁基醚、1,4-二氧六环、甲苯中的任一种或多种。
[0023]
优选的式1所示化合物与有机溶剂的体积比为:1∶(20~50),优选的式1所示化合物与有机溶剂的体积比为:1∶40。
[0024]
优选的,铯盐添加剂分别为硫酸铯和氟化铯。
[0025]
优选的,所述反应温度为110℃~40℃。
[0026]
采用本发明的技术方案至少可以达到如下有益效果之一:
[0027]
本发明直接通过甲苯去质子一锅法合成2-芳基苯乙酮类化合物,不需要制备格式试剂或是苄基金属试剂,也不需要使用过度金属催化剂。
[0028]
本发明实验操作步骤简单,无需极端的升温或降温,反应条件温和,丰富了反应的底物的适用性。
[0029]
本发明方法同时大量的筛选了反应条件,确定了最优反应条件,进一步提高反应收率。
[0030]
本发明所采用的的原料价格便宜,容易获得,反应易于操作和控制。
附图说明
[0031]
图1a、2a、3a分别为实施例1~3产物的核磁共振氢谱图,1b、2b、3b分别为实施例 1~3产物的核磁共振碳谱图
具体实施方式
[0032]
为了便于本领域技术人员理解,下面结合实施例对本发明的构思做进一步的说明。同时,说明书中所涉及的各种原料,均购自市场,二(三甲基硅基)氨基锂(aldrich,97%),二(三甲基硅基)氨基钾(aldrich,95%),硫酸铯和氟化铯(安耐吉,99%),其他药品等购自sigma-aldrich, acros,alfa aesar或者j&k.,核磁共振谱仪型号为布鲁克400兆。
[0033]
实施例1
[0034]
在手套箱内氩气气氛下放入装有搅拌磁子的烘干微波管,加入lin(sime3)2(50.2mg,0.30 mmol),cs2so4(54.3mg,0.15mmol)和干燥甲苯(0.6ml)。然后用盖子密封,从手套箱中取出。反应液在油浴加热到110摄氏度,搅拌4h。冷却到室温后,相应的苯甲酸甲酯(0.10mmol) 通过注射器加入到反应液中,然后加热到50摄氏度搅拌4h。密封微波管冷却到室温,开盖,然后加入3滴水淬灭反应。反应液过滤一下,并通过减压旋蒸浓缩,然后通过硅胶柱进行纯化。(石油醚∶乙酸乙酯=15∶1),即可得二苯乙酮(17.6mg,90%yield),产物为白色固体。产物的氢谱和碳谱核磁共振谱图分别为图1a和图1b,谱图数据为:1h nmr(400mhz,cdcl3) δ:8.05(d,j=7.4,2h),7.63-7.55(m,1h),7.49(dd,j=8.4,6.9,2h),7.41-7.33(m,2h),7.30(d, j=10.2,3h),4.32(s,2h).
13
c{1h}nmr(101mhz,cdcl3)δ:197.6,136.6,134.5,133.2,129.5, 128.69,128.66,128.63,126.9,45.5ppm.
[0035]
变换实施例1中的两种原料,设计如下3组实验实施例,其中第1组实验即为实施例1,对应的产物的核磁共振谱图为图1。其余2-3各组产物的核磁共振谱图的序号对应相应的实施例的序号。
[0036]
表中列出了1-3各实施例中r1、r2以及对应的产物的结构式,最后一列列出了各实施例的产物的产率,并标示出了各实施例的具体实施条件,各实施例实施条件的具体含义如表格下方所示。
[0037][0038]
以下为2-3各个实施例产物的1h和
13
c的核磁共振谱图(nmr)图谱数据结果。
[0039]
实施例2
[0040]
在手套箱内氩气气氛下放入装有搅拌磁子的烘干微波管,加入lin(sime3)2(50.2mg,0.30 mmol),csf(30.4mg,0.2mmol)和干燥甲苯(0.6ml)。然后用盖子密封,从手套箱中取出。反应液在油浴加热到110摄氏度,搅拌4h。冷却到室温后,相应的酰胺(0.10mmol)通过注射器加入到反应液中,然后加热到50摄氏度搅拌4h。密封微波管冷却到室温,开盖,然后加入3滴水淬灭反应。反应液过滤一下,并通过减压旋蒸浓缩,然后通过硅胶柱进行纯化。(石油醚∶乙酸乙酯=15∶1),即可得2-(4-溴苯基)苯乙酮(17.6mg,58%yield),产物为黄色固体。产物的氢谱和碳谱核磁共振谱图分别为图2a和图2b,谱图数据为:1h nmr(400mhz,cdcl3)δ: 7.87-7.84(m,2h),7.60-7.57(m,2h),7.38-7.27(m,3h),7.29-7.25(m,1h),7.23(s,2h), 4.24(s,2h).
13
c{1h}nmr(101mhz,cdcl3)δ:196.7,135.3,134.2,132.1,130.3,129.5,128.9, 128.5,127.2,45.6ppm.
[0041]
实施例3
[0042]
手套箱内氩气气氛下放入装有搅拌磁子的烘干微波管,加入lin(sime3)2(50.2mg,0.30 mmol),csf(30.4mg,0.2mmol)和干燥的2-methf(0.5ml)。然后用盖子密封,从手套箱中取出。反应液在油浴加热到110摄氏度,搅拌4h。冷却到室温后,相应的n-甲氧基-n-甲基苯甲酰胺(0.10mmol)和2,3,4,5,6-五氟甲苯通过注射器加入到反应液中,然后加热到50摄氏度搅拌4h。密封微波管冷却到室温,开盖,然后加入3滴水淬灭反应。反应液过滤,并通过减压旋蒸浓缩,然后通过硅胶柱进行纯化。(石油醚∶乙酸乙酯=15∶1),即可得到2-(五氟苯基)苯乙酮(24.9mg,87%),产物为白色固体,产物的氢谱和碳谱核磁共振谱图分别为图3a和3b,谱图数据为:1h nmr(400mhz,cdcl3)δ:8.08-7.99(m,2h),7.69-7.59(m,1h),7.56-7.49(m, 2h),4.40(s,2h).
13
c{1h}nmr(101mhz,cdcl3)δ:192.9,146.6(m),144.4(m),141.8(m), 139.0(m),136.3(m),135.7,133.9,128.9,128.3,108.5(m),32.6ppm.