1.本技术要求于2019年7月3日提交韩国专利厅的韩国专利申请第10-2019-0080188号的优先权,该专利申请的公开内容引入本说明书中作为参考。
2.本发明涉及一种作为抗流感制剂的神经氨酸酶感染抑制剂帕拉米韦三水合物的制备方法。
背景技术:
3.流感病毒(influenza virus)是一种引起急性呼吸道疾病的传染性病毒病,是一种俗称流感的病毒。1931年,shope首次从猪中分离出流感病毒,smith、andrews和laidlow于1933年从人类中分离出流感病毒。目前,流感病毒通过多种变异在世界范围内引起人类的各种急性呼吸道疾病,并因此作为一种增加发病率和死亡率的病毒进行管理。最近,当流感病毒通过各种变异出现新病毒时,预计发病率和死亡率将大幅增加。由于认识到流感病毒的重要性和严重性,世界各地都在运行流感监测系统。
4.流感病毒是属于正粘病毒科(orthomyxoviridae)的rna病毒,呈多态性球形,由8个切片组成,直径约80~120nm。在其包膜中存在两种糖蛋白,即血凝素(hemagglutinin;ha)蛋白和神经氨酸酶(neuraminidase;na)以及基质蛋白(matrix,m2),在包膜内侧界面存在另一种基质(m1)蛋白。流感病毒根据其抗原类型分为a型、b型和c型。a型流感按表面抗原ha和na确定为亚型(subtype),ha是起到病毒附着体细胞的作用,存在h1到h18的亚型。另外,na在病毒从感染细胞释放和传播到新的呼吸道细胞的过程中起重要作用,存在n1到n11的亚型。相反,b型流感病毒的抗原变化比a型小,并且具有免疫学上稳定的形式。并且,如上所述,只有人类作为唯一宿主起作用,分为维多利亚(victoria)系和山形(yamagata)系。最后,c型流感大多无症状,未知与流感流行有关联。
5.作为预防和抑制这种流感病毒增殖的治疗剂,被开发的抗流感感染抑制剂有奥司他韦(oseltamivir;tamiflu
tm
)、扎那米韦(zanamivir;rerenza
tm
)以及帕拉米韦(peramivir;peramiflu
tm
),这些均对a型及b型流感具有抗病毒效果。相反,m2抑制剂金刚烷胺(amantadine)和金刚乙胺(rimantadine)仅对a型流感病毒有效,由于耐性,目前不推荐使用。a型及b型流感病毒感染抑制剂奥司他韦,其通过阻断病毒增殖酶的功能,表现出,对流感病毒的效果,应在出现症状后48小时内服用,才能获得最大效果。然而,它的缺点是即使在症状改善后也必须服用5天,近来服用奥司他韦后,因呕吐、恶心、神经精神异常等引起的副作用严重,已成为社会问题。扎那米韦是另一种a型和b型流感病毒感染抑制剂,是一种非口服而是口腔喷洒的病毒治疗剂,它也抑制存在于病毒表面的增殖酶神经氨酸酶,并抑制病毒向其他细胞的传播,因而已知其对各种变异流感类病毒均有效的治疗剂。然而,扎那米韦也有腹泻、恶心、呕吐、头痛、头晕、流鼻血等严重副作用,并且存在只能给7岁以上的人给药的缺点。它们的全球适销性是因为扎那米韦的市场正在被蚕食,而奥司他韦也有严重的神经精神异常等严重的副作用,难以为难以口服的患者开处方,5天内必须口服10次左右,因而预计奥司他韦的市场也很可能被蚕食。相反,帕拉米韦(peramivir)是最近开发的
一类神经氨酸酶抑制剂,已知其为型和a型及b型流感病毒感染的治疗剂。就帕拉米韦而言,它具有抑制多种流感病毒株作用的作用机制,并具有可以开出单次静脉注射处方的优点。由于帕拉米韦的静脉注射处方,与口服奥司他韦和吸入扎那米韦相比,它被评估为一种非常有效的治疗剂,也可用于口服困难、代谢性疾病、慢性呼吸系统疾病的患者。并且,与口服用处方相比,具有解热快、呕吐、恶心等副作用相对较少的优点。帕拉米韦作为流感感染抑制剂,在2014年获得fda批准后,通过改善其低认知度而呈现持续增长,而在2018年9月,由于在韩国适应症的扩大,需求层逐渐增加。
6.帕拉米韦的产品名称为peramiflu(peramiflu
tm
),其制备成具有由以下化学式1表示的帕拉米韦水合物形式的注射剂。更加详细地,peramiflu具有由化学式2表示的帕拉米韦三水合物的形式。
7.化学式1
[0008][0009]
(在上述化学式中,x=1、2、3)
[0010]
化学式2
[0011][0012]
帕拉米韦三水合物的制备方法的专利通过原开发者的在先专利kr1020017016653/us2000016013/wo200100571,以及在中国申请的授权专利kr1020107005427/cn2008001459/wo2009021404及kr1020127025551/cn2008001459/wo2009021404公开。
[0013]
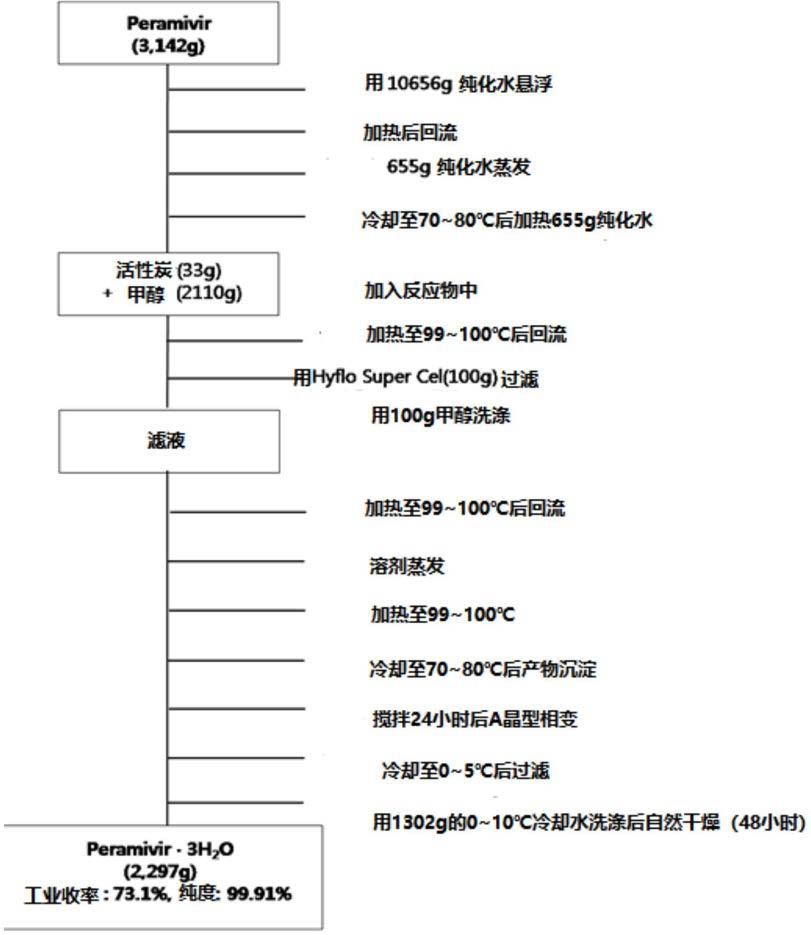
上述专利kr1020017016653/us2000016013/wo200100571中公开的帕拉米韦三水合物的制备方法如下制备工艺图1所示,kr1020107005427/cn2008001459/wo2009021404以及kr1020127025551/cn2008001459/wo2009021404的三水合物制备方法在以下制备工艺图2中更加详细地示出。
[0014]
制备工艺图1
[0015][0016]
制备工艺图2
[0017][0018]
如制备工艺图1所示,在原开发者公开的kr1020017016653/us2000016013/wo200100571专利中,通过在需要管制活性炭使用和残留量的溶剂中使用具有与药品溶剂第2类对应的固有毒性的甲醇来制备帕拉米韦三水合物,并且在制备帕拉米韦水合物后干燥成三水合物的过程中,使用自然干燥法,因而其通过不适合工业生产应用和制备优良药品及质量管理标准(以下简称gmp)的干燥工艺制备。
[0019]
本发明人通过实施干燥器中无水系状态下的真空干燥而不是自然干燥,结果可以确认接近酸酐而不是三水合物的水分值,从而推测现有专利的制备方法选择自然干燥是因为这种原因而致。但是,由于帕拉米韦三水合物是一种制备成注射剂的药品,在自然干燥中,根据内部湿度有不同的干燥条件,难以保持均匀的水分值,因此自然干燥法不适合工业化批量生产及gmp标准。
[0020]
并且,制备工艺图2所示的kr1020107005427/cn2008001459/wo2009021404以及kr1020127025551/cn2008001459/wo2009021404专利也同样使用甲醇溶剂制备三水合物,
干燥工艺也通过不适合gmp法规及工业生产的工艺制备帕拉米韦三水合物。并且,制备工艺图1及2均在活性炭存在下制备三水合物。这些专利从帕拉米韦到帕拉米韦三水合物的工业收率分别为73.1%和90%,能够分别以99.91%和99.86%的纯度制备,通过这些专利制备的产物具有14%范围内的水分值。
[0021]
因此,本发明人进行了深入的研究,以努力开发一种在制备帕拉米韦三水合物时不使用活性炭及甲醇等有毒溶剂,可以制备优良的药品,还适合质量管理标准(gmp)的制备方法。
技术实现要素:
[0022]
技术问题
[0023]
本发明提供一种在制备成注射剂的帕拉米韦三水合物的制备中遏制使用甲醇和排除使用活性炭的三水合物制备技术,上述甲醇属于需要管制残留量的药品溶剂第2类并且具有固有毒性。
[0024]
本发明的目的在于,提供一种制备成具有低毒性及比现有技术提高的合成收率的如上述化学式2所示的三水合物,在帕拉米韦三水合物的制备中,使用属于药品溶剂第3类的1-丙醇、2-丙醇、1-戊醇、2-丁醇、3-甲基-1-丁醇、2-甲基-1-丙醇的醇类溶剂。
[0025]
本发明的另一目的在于,提供一种如上述化学式2所示的帕拉米韦三水合物的制备方法,上述帕拉米韦三水合物通过易于工业生产、适合制备优良药品及质量管理标准(以下简称gmp)的干燥工艺将原料和水系一同真空干燥来制备。
[0026]
本发明的最终目的在于,根据上述制备及水系干燥工艺提供一种比现有技术(甲醇)具有更高收率及低水平残留溶剂的帕拉米韦三水合物。
[0027]
解决问题的手段
[0028]
本发明的帕拉米韦三水合物的制备工艺如以下反应式1所示。
[0029]
反应式1
[0030][0031]
本发明的帕拉米韦三水合物的制备工艺利用属于第3类的低毒性醇类溶剂来代替属于药品制备中需要管制的第2类的固有毒性甲醇溶剂。
[0032]
属于药品制备中需要管制的第2类的溶剂是指需要管制残留量的溶剂,不表现遗传毒性但在动物试验中表现出致癌性的溶剂、表现出神经毒性和致畸性等致癌性以外的不可逆毒性的溶剂及其他怀疑具有严重但可逆毒性的溶剂。
[0033]
属于药品制备中需要管制的第3类的溶剂为低毒性溶剂,其被认为对人类具有低
毒性,并且不需要处于健康原因而设置暴露极限浓度。
[0034]
根据本发明的一方面,本发明提供一种由化学式2表示的(1s,2s,3r,4r)-3-[(s)-1-乙酰氨基-2-乙基丁基]-4-胍基-2-羟基环戊烷-1-羧酸三水合物(帕拉米韦三水合物)的制备方法,
[0035]
化学式2:
[0036][0037]
上述帕拉米韦三水合物的制备方法包括:将(1s,2s,3r,4r)-3-[(s)-1-乙酰氨基-2-乙基丁基]-4-胍基-2-羟基环戊烷-1-羧酸(帕拉米韦)溶解于水中来制备水溶液的步骤;以及
[0038]
加入选自1-丙醇、2-丙醇、1-戊醇、2-丁醇、3-甲基-1-丁醇、2-甲基-1-丙醇或它们的混合物的醇类溶剂的步骤。
[0039]
在本发明的一实例中,上述帕拉米韦与水以1:1至1:30的重量比加入,具体地,以1:1至1:20、1:1至1:15、1:1至1:10、1:1至1:8、1:3至1:10、1:5至1:10、1:8至1:10、1:3至1:8或1:5至1:8的比例加入,最具体地以1:8的比例加入。当上述帕拉米韦与水的重量以1:8的比例加入时,最终合成收率最高。
[0040]
在本发明的另一实例中,上述溶解与加热一同进行。当混合帕拉米韦和水时,由于最初处于悬浮液状态,因此优选通过加热来提高溶解度。
[0041]
在本发明的具体实例中,上述加热可在50℃至100℃的温度下进行,更加具体地,可在60-100℃、70-100℃、80-100℃、90-100℃、60-95℃、70-95℃、80-95℃的温度下进行,最具体地,在90℃至95℃的温度下进行。当上述加热在90℃至95℃的温度下进行时,上述帕拉米韦和水使帕拉米韦从悬浮液状态完全溶解,可以最小化蒸发的纯化水的损失。当上述加热在95℃以上的温度下进行时,由于一部分进水(纯化水)蒸发,需要补充蒸发的水(纯化水),因而不适合工业化批量生产。
[0042]
如上所述,本发明的制备方法包括加入选自1-丙醇、2-丙醇、1-戊醇、2-丁醇、3-甲基-1-丁醇、2-甲基-1-丙醇或它们的混合物的醇类溶剂的步骤。
[0043]
在本发明的制备方法中,上述反应式1步骤的醇类溶剂属于药品制备中需要管制的第3类低毒性溶剂。
[0044]
在本发明的具体实例中,当使用2-丙醇作为上述醇类溶剂时,其表现出最高的帕拉米韦三水合物的合成收率,因而优选。
[0045]
在本发明的另一实例中,相对于水的体积,以1:1-10(醇类溶剂:水)的体积比加入
上述醇类溶剂。更加具体地,相对于水的体积,以1:2-9、1:3-8、1:4-7的体积比加入上述醇类溶剂,但不限于此。最具体地,相对于水的体积,以1:5-6的体积比加入上述醇类溶剂。根据本发明的实施例,当相对于水的体积,以1:5-6的体积比加入上述醇类溶剂时,表现出帕拉米韦三水合物的提高的合成收率。
[0046]
在本发明的一实例中,上述醇类溶剂在75℃至90℃的温度下加入。在本发明的实施例中,上述醇类溶剂在85℃的温度下加入,但不限于此。
[0047]
在本发明的另一实例中,本发明的上述制备方法还包括将加入上述醇类溶剂的水溶液冷却至15℃至30℃的步骤。更加具体地,上述15-30℃的冷却温度可以是20-30℃、25-30℃、15-25℃、20-25℃,最具体地,可以是25℃。上述冷却步骤为产生帕拉米韦三水合物晶粒的步骤。根据本发明的实施例,从约40℃开始产生帕拉米韦三水合物晶粒。
[0048]
在本发明的一实例中,上述制备方法还包括在加及冷却上述醇类溶剂后,搅拌6-24小时的步骤。更加具体地,上述搅拌时间可以是6-24小时、8-24小时、10-24小时、12-24小时、6-18小时、8-18小时、10-18小时、6-12小时、8-12小时、10-12小时,最具体地,可以是12小时。上述搅拌步骤是将产生的帕拉米韦三水合物的晶体相变为a型晶体的步骤。与以往公开的执行搅拌-静置-再搅拌的制备方法相比,持续搅拌上述时间的结晶工艺省略了静置及再搅拌工艺,因而工艺更加简单,最终产生的三水合物的晶体均匀产生,由此具有显著改善结晶物的过滤性的效果。
[0049]
在本发明中,上述制备方法还可包括在进行搅拌以相变为上述a型晶体之后,冷却至1-10℃并进行搅拌的步骤。
[0050]
在本发明中,上述制备方法还包括对产生的帕拉米韦三水合物晶体进行过滤的步骤。通过上述过滤步骤产生帕拉米韦三水合物晶体饼。
[0051]
在本发明的一实例中,本发明的制备方法还包括使用醇类水溶液对通过上述过滤产生的帕拉米韦三水合物晶体饼进行洗涤的步骤。
[0052]
在本发明的具体实例中,上述醇类水溶液可以是1-丙醇水溶液、2-丙醇水溶液、1-戊醇水溶液、2-丁醇水溶液、3-甲基-1-丁醇水溶液及2-甲基-1-丙醇水溶液。
[0053]
在本发明的再一具体实例中,上述醇类水溶液的浓度为1-30%、1-20%、1-10%、5-30%、5-20%、5-10%或10%。
[0054]
在本发明的另一具体实例中,上述洗涤过程中使用的醇类水溶液的体积可以是初始帕拉米韦重量的0.1-20倍、0.1-15倍、0.1-10倍、0.1-5倍、0.1-3倍、0.1-1倍或0.1-0.5倍,最具体地,可以是0.5倍。例如,当帕拉米韦的重量为30kg时,可以使用上述洗涤用醇类水溶液15l。
[0055]
根据现有技术,帕拉米韦三水合物晶体饼在自然干燥时需要放置48小时以上,才能生产出具有14%内均匀水分的帕拉米韦三水合物。这种自然干燥方法不适合药品的工业化批量生产,是一种不适合制备优良的药品及质量管理标准(gmp)的干燥工艺。
[0056]
因此,在本发明的一实例中,本发明的帕拉米韦三水合物的制备方法还包括对上述工艺产生的帕拉米韦三水合物晶体饼进行真空干燥或减压干燥的步骤。
[0057]
在本发明的具体实例中,上述干燥在25℃至80℃的温度条件下进行,更加具体地,在25-70℃、25-60℃、25-55℃、25-50℃、25-45℃、25-40℃、30-70℃、30-60℃、30-55℃、30-50℃、30-45℃、30-40℃、35-70℃、35-60℃、35-55℃、35-50℃、35-45℃、35-40℃的温度下
进行,最具体地,在40℃的温度下进行。
[0058]
在本发明的再一具体实例中,在上述20%至80%的湿度条件下进行蒸汽干燥,更加具体地,可以在20-70%、20-65%、20-60%、30-80%、30-70%、30-65%、30-60%、30-55%、30-50%、30-45%、30-40%、40-80%、40-70%、40-60%、40-55%、40-50%、40-45%的湿度下进行蒸汽干燥。优选地,在40-60%的湿度下进行蒸汽干燥适合于恒定保持帕拉米韦三水合物的水分值,最优选地,40%适合。
[0059]
在本发明的另一具体实例中,当本发明的洗涤工艺中使用的醇类溶剂为2-丙醇时,在40℃的温度下进行干燥适合于将帕拉米韦三水合物的水分值保持在14%内,并满足2-丙醇的残留溶剂可接受标准(50mg/天,5000ppm)。
[0060]
如本发明的实施例中所证实,通过将本发明的水系和原料一同真空干燥的工序制备的帕拉米韦三水合物具有14.6%水分及显著低水平的适合的2-丙醇残留量262ppm,这在可接受标准内。
[0061]
现有技术专利kr第10-2001-7016653号、kr第10-2010-7005427号以及kr第10-2012-7025551号中,据报道使用甲醇制造三水合物方法的工业收率分别为73.1%何90%。但是,如本发明的实施例中所证实,使用2-丙醇作为本发明的醇类溶剂时的工业收率为约95%,因而表现出显着提高的帕拉米韦三水合物的合成收率。
[0062]
并且,与使用活性炭的制备工艺图1或2不同,本发明中的帕拉米韦三水合物的制备方法可以在不使用活性炭的情况下制备帕拉米韦三水合物。
[0063]
根据本发明具体实例的帕拉米韦三水合物的制备方法如制备工艺图3所示。
[0064]
制备工艺图3
[0065]
[0066]
如上所述,可知本发明的制备方法是一种既适合及易于药品的工业生产,又可以适合制备优良的药品及质量管理标准(gmp)制备的制备方法。
[0067]
发明的效果
[0068]
本发明涉及一种作为抗流感制剂的神经氨酸酶感染抑制剂帕拉米韦三水合物的制备方法,根据本发明的制备方法,无需使用剧毒的甲醇和活性炭,而通过适合gmp标准的工艺,可以高收率、稳定地制备帕拉米韦三水合物。
附图说明
[0069]
图1为本发明的帕拉米韦三水合物的制备工艺的示意图。
[0070]
图2a为本发明的实施例2中制备的帕拉米韦三水合物的液相色谱(hplc)谱图。
[0071]
图2b为本发明的实施例2中制备的帕拉米韦三水合物的液相色谱(hplc)谱图。
[0072]
图3为本发明的实施例2中制备的化合物帕拉米韦三水合物的1h-nmr谱图。
[0073]
图4为本发明的实施例2中制备的化合物帕拉米韦三水合物的
13
c-nmr谱图。
[0074]
图5为本发明的实施例2中制备的化合物帕拉米韦三水合物的液相色谱-质谱(lc-ms)谱图。
[0075]
图6为示出本发明的实施例2中制备的化合物帕拉米韦三水合物的a晶型的x-射线衍射(xrd)值的图。
[0076]
图7a为可检测溶剂的每个可接受标准的气象色谱(gc)谱图。
[0077]
图7b为本发明的实施例2中制备的化合物帕拉米韦三水合物的气象色谱(gc)谱图。
具体实施方式
[0078]
以下,将通过实施例更详细地描述本发明。这些实施例仅用于更具体地说明本发明,根据本发明的主旨,本发明的范围不受这些实施例的限制,这对于本发明所属技术领域的普通技术人员而言是显而易见的。
[0079]
实施例
[0080]
在本说明书全文中,除非另有说明,对于用于表示特定物质浓度的“%”而言,固体/固体为(重量/重量)%、固体/液体为(重量/体积)%、另外,液体/液体为(体积/体积)%。
[0081]
试剂及分析方法
[0082]
帕拉米韦(1s,2s,3r,4r)-3-[(s)-1-乙酰氨基-2-乙基丁基]-4-胍基-2-羟基环戊烷-1-羧酸)、纯化水、1-丙醇(1-propanol)、2-丙醇(2-propanol)、1-戊醇(1-pentanol)等未经纯化过程而使用。并且,本发明中的反应工艺的进行及纯度的确认通过使用液相色谱(hplc)等仪器来进行了分析及测量,利用核磁共振(1hnmr,
13
c nmr)、液相色谱质谱仪(hplc)、x-射线衍射仪(xrd)、水分滴定仪(karl fischer)及气象色谱(gc)对结构鉴定、晶型、水分及残留溶剂进行了分析。
[0083]
实施例1:帕拉米韦三水合物((1s,2s,3r,4r)-3-[(s)-1-乙酰氨基-2-乙基丁基]-4-胍基-2-羟基环戊烷-1-羧酸三水合物)的制备(1)
[0084][0085]
在500.0l容量反应器中加入30.0kg的(1s,2s,3r,4r)-3-[(s)-1-乙酰氨基-2-乙基丁基]-4-胍基-2-羟基环戊烷-1-羧酸(帕拉米韦;peramivir),并将其悬浮在240.0l的纯化水中。将反应物加热至约95℃来使悬浮液完全溶解后搅拌,并进行自然冷却。当反应物温度为85℃时,缓慢加入45.0l的1-丙醇(1-propanol),一边搅拌一边自然冷却至25℃。此时,在约40℃的温度下产生帕拉米韦三水合物的晶粒。当通过自然冷却来使反应器温度达到25℃时,搅拌约12小时,以使a晶型发生相变。并且,将反应器温度冷却至5℃,进一步搅拌约3小时,反应结束后过滤,用10%的1-丙醇溶剂15.0l洗涤一次。将过滤的固体饼放入内部湿度约40%的真空干燥器中,在真空干燥器底部放置纯化水碗,将固体饼在40℃的温度下干燥约12小时。干燥结束后,得到帕拉米韦三水合物(收率:95.0%、纯度:99.9%、水分:14.5%)。
[0086]1h nmr(500mhz,d2o,δ):4.30-4.27(m,2h),3.79-3.74(m,1h,-ch),2.65-2.63(m,1h,-ch),2.48-1.45(m,1h,-ch),2.16-2.12(m,1h,-ch),1.88(s,3h,-ch3),1.75-1.71(m,1h,-ch),1.41-1.36(m,3h),0.95-0.91(m,2h),0.87-0.84(t,3h,-ch3),0.82-0.79(t,3h,-ch3);
13
c nmr(500mhz,d2o,δ):181.671,173.686,155.584,75.368,55.201,54.091,50.314,49.933,43.409,34.015,22.780,21.887,20.942,12.128,11.276;lc-ms(m/z,c
15h28
n4o4):calcd for 329.21,found;329.5.
[0087]
实施例2:帕拉米韦三水合物((1s,2s,3r,4r)-3-[(s)-1-乙酰氨基-2-乙基丁基]-4-胍基-2-羟基环戊烷-1-羧酸三水合物)的制备(2)
[0088][0089]
在500.0l容量反应器中加入30.0kg的(1s,2s,3r,4r)-3-[(s)-1-乙酰氨基-2-乙基丁基]-4-胍基-2-羟基环戊烷-1-羧酸(帕拉米韦;peramivir),并将其悬浮在240.0l的纯化水中。将反应物加热至约95℃来使悬浮液完全溶解后搅拌,并进行自然冷却。当反应物温
度为85℃时,缓慢加入45.0l的2-丙醇(2-propanol),一边搅拌一边自然冷却至25℃。此时,在约40℃的温度下产生帕拉米韦三水合物的晶粒。当通过自然冷却来使反应器温度达到25℃时,搅拌约12小时,以使a晶型发生相变。并且,将反应器温度冷却至5℃,进一步搅拌约3小时,反应结束后过滤,用10%的2-丙醇溶剂15.0l洗涤一次。将过滤的固体饼放入内部湿度约40%的真空干燥器中,在真空干燥器底部放置纯化水碗,将固体饼在40℃的温度下干燥约12小时。干燥结束后,得到帕拉米韦三水合物(收率:95.5%、纯度:99.9%、水分:14.6%)。
[0090]1h nmr(500mhz,d2o,δ):4.30-4.27(m,2h),3.79-3.74(m,1h,-ch),2.65-2.63(m,1h,-ch),2.48-1.45(m,1h,-ch),2.16-2.12(m,1h,-ch),1.88(s,3h,-ch3),1.75-1.71(m,1h,-ch),1.41-1.36(m,3h),0.95-0.91(m,2h),0.87-0.84(t,3h,-ch3),0.82-0.79(t,3h,-ch3);
13
c nmr(500mhz,d2o,δ):181.671,173.686,155.584,75.368,55.201,54.091,50.314,49.933,43.409,34.015,22.780,21.887,20.942,12.128,11.276;lc-ms(m/z,c
15h28
n4o4):calcd for 329.21,found;329.5.
[0091]
下表1示出本发明的实施例2中制备的化合物帕拉米韦三水合物的a晶型的2-theta(deg)值。
[0092]
表1
[0093]
[0094]
[0095]
[0096][0097]
并且,将为了反应而加入的2-丙醇的加入量调节为40l或48l来制备帕拉米韦三水合物的结果如下(表2)。
[0098]
表2
[0099][0100]
实施例3:帕拉米韦三水合物((1s,2s,3r,4r)-3-[(s)-1-乙酰氨基-2-乙基丁基]-4-胍基-2-羟基环戊烷-1-羧酸三水合物)的制备(3)
[0101][0102]
在500.0l容量反应器中加入30.0kg的(1s,2s,3r,4r)-3-[(s)-1-乙酰氨基-2-乙
基丁基]-4-胍基-2-羟基环戊烷-1-羧酸(帕拉米韦;peramivir),并将其悬浮在240.0l的纯化水中。将反应物加热至约95℃来使悬浮液完全溶解后搅拌,并进行自然冷却。当反应物温度为85℃时,缓慢加入45.0l的1-戊醇(1-pentanol),一边搅拌一边自然冷却至25℃。此时,在约40℃的温度下产生帕拉米韦三水合物的晶粒。当通过自然冷却来使反应器温度达到25℃时,搅拌约12小时,以使a晶型发生相变。并且,将反应器温度冷却至5℃,进一步搅拌约3小时,反应结束后过滤,用10%的1-戊醇(1-pentanol)溶剂15.0l洗涤一次。将过滤的固体饼放入内部湿度约40%的真空干燥器中,在真空干燥器底部放置纯化水碗,将固体饼在40℃的温度下干燥约12小时。干燥结束后,得到帕拉米韦三水合物(收率:95.1%、纯度:99.9%、水分:14.1%)。
[0103]1h nmr(500mhz,d2o,δ):4.30-4.27(m,2h),3.79-3.74(m,1h,-ch),2.65-2.63(m,1h,-ch),2.48-1.45(m,1h,-ch),2.16-2.12(m,1h,-ch),1.88(s,3h,-ch3),1.75-1.71(m,1h,-ch),1.41-1.36(m,3h),0.95-0.91(m,2h),0.87-0.84(t,3h,-ch3),0.82-0.79(t,3h,-ch3);
13
c nmr(500mhz,d2o,δ):181.671,173.686,155.584,75.368,55.201,54.091,50.314,49.933,43.409,34.015,22.780,21.887,20.942,12.128,11.276;lc-ms(m/z,c
15h28
n4o4):calcd for 329.21,found;329.5.