1.本发明涉及一种热活化延迟荧光聚合物及其应用,可用于柔性光电有机半导体技术领域。
背景技术:
2.恶性肿瘤(癌症)已成为当前对人类健康安全危害最大的疾病之一。临床治疗中,以手术和放、化疗为主的治疗手段虽然取得了长足进展,但恶性肿瘤发病机理复杂、易复发和高转移等问题的存在,使得上述常见治疗方法尚难说成功。例如,化疗不仅存在全身毒性和多药耐药问题,也常常会损害周围的健康组织,最终影响患者的生存率。此外,恶性肿瘤之外,炎症相关的细菌和真菌感染、帕金森病等老年退行性疾病同样面临越来越少特效药物的问题。与治疗手段缺乏相比,人类生存环境变化引起的新型流行病或病毒感染对全人类的健康和生存造成极大威胁。面对险恶疾病缺乏有效的早期检测、预防手段,也没有针对性特效药物,迫切需要科研工作者重新审视重大疾病检测、诊断和治疗方案的研究思路。
3.在各种新兴的癌症治疗手段中,光动力疗法(photodynamic therapy,pdt)具有无创性、副作用少、耐药性小、全身毒性低和可与多种其他疗法联合使用等优点而受到越来越多的关注。典型的pdt过程,需要三个关键要素:光敏剂(photosensitizer,ps)、能激发光敏剂的光源和组织氧。无毒的ps受光源激发向组织氧发生电荷转移(i型光动力)或能量转移(ii型光动力)产生具有细胞毒性的活性氧(ros,典型的如单线态氧 (1o2))。pdt杀死肿瘤细胞的途径包括凋亡、坏死、刺激宿主免疫系统,引起急性炎症和白细胞浸润肿瘤(增加肿瘤源性抗原对t细胞的表达)。
4.光敏剂是光动力治疗的关键。经过至少两代ps的迭代发展,已有数千种pss被发展并应用于靶向抗菌、抗肿瘤、眼科、痤疮、血管等多种疾病病的治疗。pdt的研究关键是发展高效的光敏剂。最有效的光敏剂具有以下特征:1)s1向t1的系间窜越速率常数高;2)光稳定性好;3)近红外区光吸收能力强;4)在肿瘤细胞中有高选择性和高肿瘤倾向性,保证其具有足够好的肿瘤细胞毒性,同时,光敏剂暗毒性要低、易于组织或人体清除。光敏剂按照材料结构和组成分为:1)含重金属的ps(潜在的元素毒性);2)贵金属纳米材料(密度和成本高);3)全有机小分子ps和4)聚合物ps(包括共轭聚电解质、有机金属框架(mofs)、共价有机框架(cofs)等)。相对而言,全有机ps 的生物安全性更好、也更容易通过化学结构修饰进行功能化。目前,全有机ps主要是卟啉衍生物、卟吩衍生物、氟硼二吡咯(bodipy)衍生物、吲哚菁(ic)衍生物、吡咯并吡咯二酮(dpp)衍生物。虽然,这几类共轭有机半导体单元,能很方便的通过偶联反应和其他化学修饰获得高效的ps,但更多的时候,共轭功能化会导致基于以上结构的化合物失去其pdt性质。例如,dpp偶联三苯胺或噻吩并吲哚单元之后,具有良好的光热转换效率,但不具有pdt能力。有机小分子ps在构建抗肿瘤诊疗试剂时,还容易从纳米包覆体系中渗漏、聚集引起荧光猝灭和光动力效能丧失等问题。
5.无论从光动力治疗和诊疗一体化哪个方面看,发展高效光敏剂和近红外纳米探针对生物医学应用都具有非常重要的理论意义和市场价值。
技术实现要素:
6.本发明的目的就是为了解决现有技术中存在的上述问题,提出一种热活化延迟荧光聚合物及其应用。
7.本发明的目的将通过以下技术方案得以实现:一种热活化延迟荧光聚合物,该热活化延迟荧光聚合物的结构式为:
[0008][0009]
通式(i)
[0010]
其中,通式(i)中,x表示苯环a和苯环b之间的连接方式,可以是苯环a与苯环 b没有经过x的连接,可以是单键相连、经过-nr
1-、-o-和-s-中的任意一种;
[0011]
m+n=0.5,且0.5>m>0;ar1为取代的苯、萘、芴、咔唑、螺芴、蒽、氧杂蒽,ar1 上的取代为c1~c20直链取代基、c1~c20的支链取代基、c1~c20直链烷氧基或c1~c20 支链烷氧基中的一种;
[0012]
ar2取自取代的苯、萘、芴、咔唑、螺芴、蒽、氧杂蒽、苯并噻二唑苯并硒二唑吡啶并噻二唑5,6-二氟苯并噻二唑 5-氟苯并噻二唑4,7-二芳基-5,6-二烷氧基苯并噻二唑 4,7-二芳基苯并噻二唑4,7-二芳基苯并硒二唑4,7
‑ꢀ
二芳基吡啶并噻二唑4,7-二芳基-5,6-二氟苯并噻二唑或4,7-二芳基-5-氟苯并噻二唑中的一种,r3为具有6~16个碳原子的直链或支链烷基,b为噻吩、硒吩、并噻吩或苯并噻吩;ar2的取代为c1~c20直链取代基、c1~c20的支链取代基、c1~c20直链烷氧基或c1~c20支链烷氧基中的一种;
[0013]
ar3取自为取代或取代的苯、萘、吡啶、蒽中的一种,ar3上的取代为c1~c8的支链
或支链烷基中的一种。
[0014]
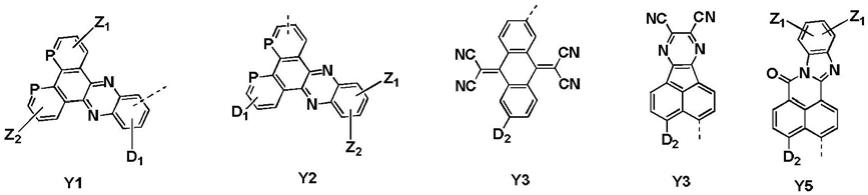
优选地,y取自以下y1、y2、y3、y4、y5中的一种:
[0015][0016]
y1~y5中:z1和z2各自独立选自氰基、氟原子和三氟甲基;p选自-ch=或n。
[0017]
优选地,d1和d2各自独立选自氢、氘、氰基、氟、三氟甲基、c1-c10的烷基、c2-c10的烯基、取代或未取代c1-c10的烷氧基、取代或未取代c6-c30的芳基、取代或未取代c3-c30的杂芳基、l-nar1ar2中的一种;l选自单键、取代或未取代c6-c30 的芳基、取代或未取代的c3-c30的杂芳基。
[0018]
优选地,ar1、ar2各自独立选自取代或未取代c6-c30的芳基、取代或未取代的c3-c30 的杂芳基;ar1和ar2彼此不连接,或ar1和ar2对经单键、-o-,-s-,-c(ch3)
2-,-c(c6h5)2‑ꢀ
连接成更大的共轭单元。
[0019]
优选地,所述ar1和ar2选自如下基团中的任意一种:
[0020][0021]
其中,虚线代表基团的连接位点;
[0022]y11
、y
12
、y
13
、y
14
、y
15
、y
16
、y
17
、y
18
、y
19
、y
110
、y
111
、y
112
各自独立地选自n或c-ry;
[0023]
t1选自o、s、n-r
t1
、cr
t2rt3
、sir
t2rt3
;
[0024]ry
、r
t1
、r
t2
、r
t3
各自独立地选自氢、氘、氰基、卤素、取代或未取代的c1~c4直链或支链烷基、取代或未取代的c6~c18芳基、取代或未取代c3~c18杂芳基、 c6~c18芳胺基;取代基ry彼此不连接或相邻的至少2个取代基通过化学键连接成环。
[0025]
优选地,所述ar选自如下基团中的任意一种,或被取代基取代的如下基团中的任意一种:
[0026][0027]
所述取代基选自氘、氟、氯、氰基、未取代或r
′
取代的c1~c4直链或支链烷基。
[0028]
本发明还揭示了一种热活化延迟荧光聚合物的应用,该热活化延迟荧光聚合物可作为光动力治疗的光敏剂和生物成像的纳米探针组分使用。
[0029]
优选地,该光动力治疗和生物成像可对肿瘤治疗、抗菌治疗和其他疾病治疗和保健应用。
[0030]
优选地,该光动力治疗的光敏剂为权利要求1~8任意一项所述的一种热活化延迟荧光聚合物,其为具有结构通式(i)的结构,光动力治疗的光敏剂的结构通式中所有标示的x、ar1、ar2、ar3、y与为权利要求1~8的限制一致。
[0031]
优选地,该热活化延迟荧光聚合物,通过制备纳米试剂来实现;该纳米试剂制备方法包括再沉淀法、分子胶束载体、以及薄膜分散法、回流沉淀法等常用有机半导体纳米试剂制备方法来实现。
[0032]
本发明采用以上技术方案与现有技术相比,具有以下技术效果:本技术方案通过简单的合成手段获得高荧光量子效率、发射波长较长、光动力活性高等优点,作为生物医学应用的纳米试剂的活性组分使用。
[0033]
该技术方案具有近红外发光、高荧光量子产率、更好的光动力治疗活性,同时给出了其生物医学应用,该应用为光动力治疗和生物成像。该热活化延迟荧光聚合物作为光动力治疗的光敏剂和荧光成像纳米试剂的关键活性组分,既能光动力治疗的光敏剂,又能充当近红外成像的荧光探针,可实现无损、光化学诊疗一体化,适合在产业上推广应用。
[0034]
本技术方案在不使用重原子和金属元素,获得柔性聚合物,实现光动力和荧光成像,为柔性电子在生物医学应用,提供了一种可行的思路。
[0035]
本发明提供的诊疗一体化纳米试剂制备方法简单、便于操作、环境稳定性好、易于负载药物和靶向处理,显著提升了其诊疗效能。本发明通过结构设计和化学合成,从聚合物分子角度实现诊疗一体化纳米试剂制备与应用,具有非常明确的应用环境和市场价值。
附图说明
[0036]
图1为本发明实施例1中聚合物a的合成路线图。
[0037]
图2为本发明实施例2中聚合物b的合成路线图。
[0038]
图3为本发明实施例3中聚合物c的合成路线图。
[0039]
图4为本发明实施例4中聚合物d的合成路线图。
[0040]
图5为本发明实施例5中聚合物e的合成路线图。
[0041]
图6为本发明实施例6中聚合物f的合成路线图。
[0042]
图7为本发明实施例3中共轭聚合物紫外吸收光谱图。
[0043]
图8为本发明实施例3中共轭聚合物荧光发射光谱图。
[0044]
图9为基于本发明实施例7中共轭聚合物的诊疗一体化纳米试剂的紫外吸收光谱图。
[0045]
图10为基于本发明实施例7中共轭聚合物的诊疗一体化纳米试剂的荧光光谱图。
[0046]
图11为施例7的热活化延迟荧光共轭聚合物的光动力/荧光诊疗一体化纳米试剂的结构示意图。
[0047]
图12为本发明实施例7中诊疗一体化纳米试剂的光动力曲线测试图。
具体实施方式
[0048]
本发明的目的、优点和特点,将通过下面优选实施例的非限制性说明进行图示和解释。这些实施例仅是应用本发明技术方案的典型范例,凡采取等同替换或者等效变换而形成的技术方案,均落在本发明要求保护的范围之内。
[0049]
本发明揭示了一种热活化延迟荧光聚合物及其应用,该热活化延迟荧光聚合物是基于主链三元共聚合骨架作为自掺杂主体构建,骨架也作为热活化延迟荧光发光体给体单元使用。一种热活化延迟荧光共轭聚合物的应用为光动力治疗和生物成像。
[0050]
本发明所用原料为已知化合物,可在市场上购得,或可用本领域已知方法合成。
[0051]
一种热活化延迟荧光共轭聚合物,所述共轭聚合物的结构式为:
[0052][0053]
通式(i)
[0054]
其中,通式(i)中,x表示苯环a和苯环b之间的连接方式,可以是苯环a与苯环 b没有经过x的连接,可以是单键相连、经过-nr
1-、-o-和-s-中的任意一种。
[0055]
通式(i)中m+n=0.5,且0.5>m>0;ar1为取代的苯、萘、芴、咔唑、螺芴、蒽、氧杂蒽,ar1的取代为c1~c20直链取代基、c1~c20的支链取代基、c1~c20直链烷氧基或c1~c20支链烷氧基中的一种。
[0056]
ar2取自取代的苯、萘、芴、咔唑、螺芴、蒽、氧杂蒽、苯并噻二唑苯并硒
二唑吡啶并噻二唑5,6-二氟苯并噻二唑 5-氟苯并噻二唑4,7-二芳基-5,6-二烷氧基苯并噻二唑 4,7-二芳基苯并噻二唑4,7-二芳基苯并硒二唑4,7
‑ꢀ
二芳基吡啶并噻二唑4,7-二芳基-5,6-二氟苯并噻二唑或4,7-二芳基-5-氟苯并噻二唑中的一种,r3为具有6~16个碳原子的直链或支链烷基,b为噻吩、硒吩、并噻吩或苯并噻吩;ar2上的取代为为c1~c20直链取代基、c1~c20的支链取代基、c1~c20直链烷氧基或c1~c20支链烷氧基中的一种。
[0057]
ar3取自为取代或取代的苯、萘、吡啶、蒽中的一种,ar3上的取代为c1~c8的支链或支链烷基中的一种。
[0058]
y取自以下y1、y2、y3、y4、y5中的一种:
[0059][0060]
y1~y5中:z1和z2各自独立选自氰基、氟原子和三氟甲基;p选自-ch=或n; d1和d2各自独立选自氢、氘、氰基、氟、三氟甲基、c1-c10的烷基、c2-c10的烯基、取代或未取代c1-c10的烷氧基、取代或未取代c6-c30的芳基、取代或未取代c3-c30 的杂芳基、l-nar1ar2中的一种;l选自单键、取代或未取代c6-c30的芳基、取代或未取代的c3-c30的杂芳基。
[0061]
ar1、ar2各自独立选自取代或未取代c6-c30的芳基、取代或未取代的c3-c30的杂芳基;ar1和ar2彼此不连接,或ar1和ar2对经单键、-o-,-s-,-c(ch3)
2-,-c(c6h5)2‑ꢀ
连接成更大的共轭单元。
[0062]
所述ar1和ar2选自如下基团中的任意一种:
[0063][0064]
其中,虚线代表基团的连接位点;y
11
、y
12
、y
13
、y
14
、y
15
、y
16
、y
17
、y
18
、 y
19
、y
110
、y
111
、y
112
各自独立地选自n或c-ry。
[0065]
t1选自o、s、n-r
t1
、cr
t2rt3
、sir
t2rt3
;ry、r
t1
、r
t2
、r
t3
各自独立地选自氢、氘、氰基、卤素、取代或未取代的c1~c4直链或支链烷基、取代或未取代的c6~c18芳基、取代或未取代c3~c18杂芳基、c6~c18芳胺基;取代基ry彼此不连接或相邻的至少2个取代基通过化学键连接成环。
[0066]
所述ar选自如下基团中的任意一种,或被取代基取代的如下基团中的任意一种:
[0067][0068]
所述取代基选自氘、氟、氯、氰基、未取代或r
′
取代的c1~c4直链或支链烷基。
[0069]
本发明还提供了一种热活化延迟荧光聚合物的应用,该应用为将一种热活化延迟荧光聚合物作为荧光发光体和光动力治疗的光敏剂,构建诊疗一体化纳米试剂。
[0070]
利用两亲聚合物自组装包裹本项目制备的新型热活化延迟荧光分子,可同时将药物分子、靶向识别单元引入纳米探针体系。此方法具有制备时间短、重复性好、颗粒粒径易于控制等非常适用于有机半导体纳米探针的制备。
[0071]
基于一种热活化延迟荧光聚合物构建的诊疗一体化纳米试剂的制备方法包括:再沉淀法、分子胶束载体、以及薄膜分散法、回流沉淀法等常用有机半导体纳米试剂制备方法来实现。
[0072]
再沉淀法制备一种活化延迟荧光聚合物的纳米试剂的方法为:首先将有机半导体和功能化包覆剂(如psma或dspe-peg2000等)分别溶解于四氢呋喃(thf),配制成浓度为1mg/ml的溶液。经过惰性气体保护条件下搅拌过夜后,以7μm的玻璃纤维滤器对包覆剂溶液进行过滤,以除去浴液中的不可溶物质。然后,将以上两种溶液按一定比例进行混合。
[0073]
例如混合溶液中1mg/ml的有机半导体和0.2mg/ml的psma。为了调控有机点粒径尺寸,将使用不同含水量的四氢呋喃(含水体积分数0%、30%、50%等),对上述混合溶液分别地进行稀释。稀释后的前驱体溶液各各组分浓度分别为50μg/ml(有机半导体) 和10μg/ml
(psma)。随后在超声水浴的条件下,将5ml稀释的前驱体聚合物混合溶液快速地注入10ml去离子水中,并持续超声2min。氮气保护下对获得的悬浮溶液加热处理,并最终将溶液浓缩至5ml左右。最后,使用0.2μm的水系薄膜过滤器对有机点分散系进行过滤。
[0074]
通过以上操作方案所制备的聚合物纳米颗粒悬浮浴液可以保持长期的稳定状态,即便是在存放数月之后仍观察不到明显的聚集现象。
[0075]
薄膜分散法制备一种活化延迟荧光聚合物的纳米试剂的方法为:将合成的荧光分子与功能双亲聚合物利用憎水溶剂氯仿(二氯甲烷等)制备成有机混合溶液,在减压蒸发仪上进行减压蒸发旋去溶剂,在瓶壁上形成均匀薄膜,再将超纯水置于上述瓶中进行超声处理,可得到水相可分散的荧光纳米探针。此方法能非常方便的在纳米颗粒中复杂更为复杂的组分,并形成非常稳定的纳米颗粒。此外,两亲分子对荧光分子聚集体的包裹,能有效改善纳米探针的界面性质,有利的保持其光学特性。
[0076]
回流沉淀法制备一种活化延迟荧光聚合物的纳米试剂的方法为:首先将本项目合成的含有羟基、氨基的荧光分子进行甲基丙烯酰化处理,获得含有一个或多个丙烯酰基的荧光分子。将此荧光分子与丙烯酸、异丙基丙烯酰胺等进行回流沉淀法聚合,可制备形貌均已的含荧光分子的功能化纳米球。回流沉淀法制备的纳米探针不仅形貌均一、且可将温敏、ph敏感基团引入其中,有利于后期进行药物吸附负载、化学键键合rgd等靶向识别基团。)
[0077]
基于一种热活化延迟荧光聚合物的诊疗一体化纳米试剂的生物医学应用,可将所述诊疗一体化纳米试剂对活体或细胞、组织进行注射、喷雾等,在适合的光照或荧光仪的匹配下,实现光动力治疗和荧光成像。
[0078]
本发明将一种热活化延迟荧光共轭聚合物应用于诊疗一体化纳米试剂,将此共轭聚合物的近红外发光特性、高荧光量子产率、高的光动力活性和高摩尔吸管吸收综合起来,获得荧光性质稳定、环境耐受性强、荧光信号好、光动力治疗效果好的诊疗一体化纳米试剂,既能对活体进行科学研究,也能作为纳米试剂进行临床环境的医学应用。这种热活化延迟荧光聚合物的主链刚性强、骨架中共轭单元分布合理,能有效保障聚合物堆积过程中优秀光物理性质的保持,为基于热活化延迟荧光发光体的生物医学应用,提供新的思路和范例。
[0079]
该热活化延迟荧光聚合物可作为光动力治疗的光敏剂和生物成像的纳米探针组分使用。该热活化延迟荧光聚合物,通过制备纳米试剂来实现的。该纳米试剂制备方法包括再沉淀法、分子胶束载体、以及薄膜分散法、回流沉淀法等常用有机半导体纳米试剂制备方法来实现。该热活化延迟荧光聚合物的光动力治疗和生物成像可对肿瘤治疗、抗菌治疗和其他疾病治疗和保健应用。
[0080]
实施例1
[0081]
本实施例提供了一种基于热活化延迟荧光共轭聚合物a,共轭聚合物a的结构式如下:
[0082][0083]
通式a
[0084]
共轭聚合物的合成路线如通式a所示。
[0085]
取0.40g(0.57mmol)2,2
’‑
(2-(4-(双(4-溴苯基)氨基)苯基)蒽-9,10-二亚烷基)二甲基腈,0.28g(1.42mmol)(2,5-二甲基-1,4-亚苯基)二硼酸和0.47g(0.85 mmol)2,7-二溴-9,9-二辛基-9h-芴加入到50ml反应管中,然后向反应管中依次加入 2mcs2co3(aq)(1ml),9.2mg(0.008mmol)催化剂四三苯基膦钯,5.3mg(0.013mmol) 配体甲基三辛基氯化铵,5ml无水甲苯,在95℃以及氩气保护条件下,搅拌反应96h,得到聚合物。将聚合物冷却至室温后,慢慢倒入150ml甲醇中,形成沉淀,沉淀的聚合物过滤后在索式提取器中依次用甲醇、正己烷来洗涤,最后用三氯甲烷溶解后沉淀到甲醇中,过滤,100℃真空干燥12h得到紫红色的固体粉末聚合物,产率49%,通式a 的结构式如图1所示。
[0086]
实施例2
[0087]
本实施例提供了一种基于多环芳烃的共轭聚合物b,共轭聚合物的结构通式b如下:
[0088][0089]
通式b
[0090]
共轭聚合物的合成路线如通式b所示。
[0091]
取0.50g(0.73mmol)2,2
’‑
(2-(4-(双(4-溴苯基)氨基)苯基)蒽-9,10-二亚烷基)二甲基腈,1.17g(2.44mmol)(9,9-二辛基-9h-芴-2,7-二基)二硼酸和0.96 g(1.71mmol)2,
7-二溴9-(十七烷-9-基)-9h-咔唑加入到50ml反应管中,反应管中依次加入2mcs2co3(aq)(1ml),9.2mg(0.008mmol)催化剂四三苯基膦钯,5.3mg (0.013mmol)配体甲基三辛基氯化铵,5ml无水甲苯,在95℃以及氩气保护条件下,搅拌反应96h,得到聚合物。将聚合物冷却至室温后,慢慢倒入150ml甲醇中,形成沉淀,沉淀的聚合物过滤后在索式提取器中依次用甲醇、正己烷来洗涤,最后用三氯甲烷溶解后沉淀到甲醇中,过滤,100℃真空干燥12h得到紫黑色的固体粉末聚合物,产率57%,通式b的结构式如图2所示。。
[0092]
实施例3
[0093]
本实施例提供了一种基于多环芳烃的共轭聚合物c,共轭聚合物c的结构通式c如下:
[0094][0095]
通式c
[0096]
共轭聚合物的合成路线如通式c所示。
[0097]
取0.2g(0.24mmol)(4-(7-(4-(双(4-溴苯基)氨基)苯基)-10,13-二氰基二苯并[a,c]吩嗪-2-基)-3,5-二甲基苯基),0.46g(1.18mmol)(9-(十七烷-9-基)-9h
‑ꢀ
咔唑-3,6-二基)二硼酸和0.52g(0.94mmol)2,7-二溴-9,9-二辛基-9h-芴加入到50ml 反应管中,然后向反应管中依次加入2mcs2co3(aq)(1ml),9.2mg(0.008mmol)催化剂四三苯基膦钯,5.3mg(0.013mmol)配体甲基三辛基氯化铵,5ml无水甲苯在95℃以及氩气保护条件下,搅拌反应96h,得到聚合物。将聚合物冷却至室温后,慢慢倒入 150ml甲醇中,形成沉淀,沉淀的聚合物过滤后在索式提取器中依次用甲醇、正己烷来洗涤,最后用三氯甲烷溶解后沉淀到甲醇中,过滤,100℃真空干燥12h得到紫黑色的固体粉末聚合物,产率46%,通式c的结构式如图3所示。
[0098]
实施例4
[0099]
本实施例提供了一种基于多环芳烃的共轭聚合物d,共轭聚合物d的结构通式d如下:
[0100][0101]
通式d
[0102]
共轭聚合物的合成路线如通式d所示。
[0103]
取0.4g(0.41mmol)2-(4-(双(4-溴苯基)氨基)苯基)-7-(4-(二苯基氨基) 苯基)二苯并[a,c]吩嗪-10,13-二甲腈,0.51g(1.02mmol)(9-(十七烷-9-基)-9h
‑ꢀ
咔唑-2,7-二基)二硼酸和0.16g(0.62mmol)1,4-二溴-2,5-二甲苯加入到50ml反应管中,然后向反应管中依次加入2mcs2co3(aq)(1ml),9.2mg(0.008mmol)催化剂四三苯基膦钯,5.3mg(0.013mmol)配体甲基三辛基氯化铵,5ml无水甲苯在95℃以及氩气保护条件下,搅拌反应96h,得到聚合物。将聚合物冷却至室温后,慢慢倒入 150ml甲醇中,形成沉淀,沉淀的聚合物过滤后在索式提取器中依次用甲醇、正己烷来洗涤,最后用三氯甲烷溶解后沉淀到甲醇中,过滤,100℃真空干燥12h得到紫黑色的固体粉末聚合物,产率53%,,通式e的结构式如图4所示。
[0104]
实施例5
[0105]
本实施例提供了一种基于多环芳烃的共轭聚合物e,共轭聚合物e的结构通式e如下:
[0106][0107]
通式e
[0108]
共轭聚合物的合成路线如通式e所示。
[0109]
取0.30g(0.43mmol)2,2
’‑
(2-(4-(4-(3,6-二溴-9h-咔唑-9-基)苯基)蒽-9, 10-二亚烷基)二甲基腈,0.70g(1.43mmol)(9-(十七烷-9-基)-9h-咔唑-3,6-二基) 二硼酸和0.55g(1.00mmol)2,7-二溴-9,9-二辛基-9h-芴加入到50ml反应管中,然后向反应管中依次加入2mcs2co3(aq)(1ml),9.2mg(0.008mmol)催化剂四三苯基膦钯,5.3mg(0.013mmol)配体甲基三辛基氯化铵,5ml无水甲苯在95℃以及氩气保护条件下,搅拌反应96h,得到聚合物。将聚合物冷却至室温后,慢慢倒入150ml甲醇中,形成沉淀,沉淀的聚合物过滤后在索式提取器中依次用甲醇、正己烷来洗涤,最后用三氯甲烷溶解后沉淀到甲醇中,过滤,100℃真空干燥12h得到紫红色的固体粉末聚合物,产率为56%,通式f的结构式如图5所示。
[0110]
实施例6
[0111]
本实施例提供了一种基于多环芳烃的共轭聚合物f,共轭聚合物f的结构通式f如下:
[0112][0113]
通式f共轭聚合物的合成路线如图6所示。
[0114]
取0.20g(0.28mmol)2,2
’‑
(2-(4-(双(4-溴苯基)氨基)苯基)蒽-9,10-二亚烷基)二甲基腈,0.70g(1.41mmol)((9-(十七烷-9-基)-9h-咔唑-3,6-二基)二硼酸和0.64g(1.13mmol)2,7-二溴9-(十七烷-9-基)-9h-咔唑加入到50ml反应管中,然后向反应管中依次加入2mcs2c03(aq)(1ml),9.2mg(0.008mmol)催化剂四三苯基膦钯,5.3mg(0.013mmol)配体甲基三辛基氯化铵,5ml无水甲苯,在95℃以及氩气保护条件下,搅拌反应96h,得到聚合物。将聚合物冷却至室温后,慢慢倒入150ml 甲醇中,形成沉淀,沉淀的聚合物过滤后在索式提取器中依次用甲醇、正己烷来洗涤,最后用三氯甲烷溶解后沉淀到甲醇中,过滤,100℃真空干燥12h得到紫红色的固体粉末聚合物,产率54%,通式f的结构式如图6所示。
[0115]
实施例7
[0116]
本实施例提供了一种基于共轭聚合物制备诊疗一体化纳米试剂制。
[0117]
该诊疗一体化纳米试剂制备步骤如下:首先将实例3中的聚合物c和功能化包覆剂 (如psma或dspe-peg2000等)分别溶解于四氢呋喃(thf),配制成浓度为1mg/ml的溶液。经过惰性气体保护条件下搅拌过夜后,以7μm的玻璃纤维滤器对包覆剂溶液进行过滤,以除去浴液中的不可溶物质;然后,将以上两种溶液按一定比例进行混合。例如混合溶液中1mg/ml的有机半导体和0.2mg/ml的psma。
[0118]
为了调控有机点粒径尺寸,将使用不同含水量的四氢呋喃(含水体积分数0%、30%、50%等),对上述混合溶液分别地进行稀释。稀释后的前驱体溶液各各组分浓度分别为50 μg/ml(有机半导体)和10μg/ml(psma);随后在超声水浴的条件下,将5ml稀释的前驱
体聚合物混合溶液快速地注入10ml去离子水中,并持续超声2min。氮气保护下对获得的悬浮溶液加热处理,并最终将溶液浓缩至5ml左右,最后,使用0.2μm的水系薄膜过滤器对有机点分散系进行过滤。
[0119]
通过以上操作方案所制备的聚合物纳米颗粒悬浮浴液可以保持长期的稳定状态,即便是在存放数月之后仍观察不到明显的聚集现象。
[0120]
所有测试结果表明,本实施例所涉及的一种共轭聚合物的将近红外发光特性、高荧光量子产率、高的光动力活性和高摩尔吸管吸收综合起来,获得荧光性质稳定、环境耐受性强、荧光信号好、光动力治疗效果好的诊疗一体化纳米试剂,既能对活体进行科学研究,也能作为纳米试剂进行临床环境的医学应用。
[0121]
将实施例3中制备的共轭聚合物c使用不同的溶剂(氯苯、二氯甲烷、四氢呋喃、甲苯)溶解并用紫外可见近吸收仪和荧光分光光度计进行紫外吸收和荧光发射的测试,图7为实施例3中制备的共轭聚合物c的紫外吸收光谱,横坐标为吸收波长,纵坐标为吸收强度,图中显示该聚合物的最大吸收波长大约在580nm处。图8为共轭聚合物c的荧光发射光谱,横坐标为发射波长,纵坐标为荧光发射强度,由图可知该聚合物的最大发射波长在680nm处。
[0122]
将实施例7中制备的纳米颗粒使用紫外可见近吸收仪和荧光分光光度计进行紫外吸收和荧光发射的测试,图9为紫外吸收光谱,横坐标为吸收波长,纵坐标为吸收强度,图中显示该纳米颗粒的最大吸收波长大约在560nm处。图10为该纳米颗粒的荧光发射光谱,横坐标为发射波长,纵坐标为荧光发射强度,由图可知该聚合物的最大发射波长在660nm处。
[0123]
将实施例7中制备的纳米颗粒使用透射电子显微镜(tem)进行表征。通过图11 可以看出通过施例7制备出来的纳米颗粒分散的比较均匀,同时制备出来的纳米颗粒为椭球形,粒径大小也比较相近,在水中能够均匀的分布。图11中的标尺为2μm,可以看出该方法制备的纳米颗粒的大小大约在100nm左右。
[0124]
为了评判实施例7制备的纳米颗粒产生单线态氧能力的强弱,本次实验选取9,10
‑ꢀ
蒽基-双(亚甲基)二丙二酸(abda)为单线态氧指示剂。将配制好的浓度为2m的abda 溶剂中取10μl加入适合浓度的纳米颗粒水溶液中,使用100mw
·
cm-2
的白光光源(400-800nm)进行照射。以60s为间隔记录在300s内指示剂在378nm处的吸光度的下降趋势。从图12中可以看出,当在光照的条件下abda在378nm处吸收强度随着照射时间增加而降低,说明制备出来的纳米颗粒水溶液具有光动力特性。
[0125]
本发明尚有多种实施方式,凡采用等同变换或者等效变换而形成的所有技术方案,均落在本发明的保护范围之内。