1.本发明涉及超支化聚合物领域,特别涉及一种超支化聚咪唑啉类化合物及其制备方法和应用。
背景技术:
2.超支化聚合物由于其含有大量高度支化的三维结构而表现出特殊的性质。相较于线形聚合物,超支化聚合物往往具有较低的粘度、较低的结晶度、较好的溶解性、易修饰等特点,因而获得了高分子科学家的广泛关注。现如今,超支化聚合物在涂料、药物释放、纳米科学等领域有着广泛的应用,因此,探索新的可制备超支化聚合物的聚合反应以及具有新型功能的超支化聚合物具有重要的研究意义。
3.异腈基乙酸酯类单体与磺酰亚胺单体制备线形聚咪唑啉的聚合反应已被成功报道,然而将该聚合反应用于制备超支化聚合物仍是空白。
技术实现要素:
4.为了克服现有技术的上述缺点与不足,本发明的目的在于提供一种超支化聚咪唑啉类化合物,具有良好的可加工性、高的热稳定性、可降解性。
5.本发明的另一目的在于提供一种超支化聚咪唑啉的制备方法,操作简单,反应高效。
6.本发明的再一目的在于提供一种三元异腈基化合物的应用。
7.本发明的目的通过以下技术方案实现:
8.一种超支化聚咪唑啉类化合物,具有如下结构:
[0009][0010]
其中,r1表示烷基或芳基,r2表示烷基或芳基。
[0011]
优选的,所述r1为(ii-1)~(ii-2)中的一种:
[0012][0013]
r2为(iii-1)~(iii-2)中的一种:
[0014][0015]
其中*表示取代位置。
[0016]
所述的超支化聚咪唑啉类化合物制备方法,包括以下步骤:
[0017]
将三元异腈基化合物、二元磺酰亚胺基化合物和聚合催化剂在有机溶剂中,得到反应液,经过聚合反应,在甲醇中沉淀,过滤得到超支化聚磺酰亚胺类化合物;
[0018]
其中,所述的三元异腈基化合物如式(ii)所示:
[0019][0020]
式(ii)中,r1为(ii-1)~(ii-2)中的一种:
[0021][0022]
其中,*表示取代位置。
[0023]
优选的,所述二元磺酰亚胺基化合物的结构如式(iii)所示:
[0024][0025]
式(iii)中,r2为(iii-1)~(iii-2)中的一种:
[0026][0027]
其中,*表示取代位置。
[0028]
优选的,所述三元异腈基化合物与所述的二元磺酰亚胺基化合物的摩尔比为1:
(0.8~1.2)。
[0029]
优选的,所述聚合催化剂包含氯化亚铜、三苯基膦和三乙胺;其中所述氯化亚铜与三元异腈类化合物摩尔比为(8~10):100,三苯基膦与三元异腈类化合物摩尔比为(16~20):1,三乙胺与三元异腈类化合物摩尔比为(8~10):100。
[0030]
优选的,所述反应液中三元异腈基化合物的浓度为0.05~0.2mol/l。
[0031]
优选的,所述聚合反应的温度为0~40℃;反应时间为1~8小时。
[0032]
优选的,所述有机溶剂为二氯甲烷、氯仿、四氢呋喃、n,n-二甲基甲酰胺中的至少一种。
[0033]
所述的超支化聚咪唑啉类化合物在可降解材料中的应用。
[0034]
本发明合成了多种多元异腈基乙酸酯类单体,将其与二元磺酰亚胺单体反应制备了多种超支化聚噁咪唑啉;该聚合物结构明确,可溶于有机溶剂,具有良好的热稳定性,可加工性强。此外,该类型超支化聚合物可在碱性环境下高效降解,具有环境友好性。
[0035]
与现有技术相比,本发明具有以下优点和有益效果:
[0036]
(1)本发明的超支化聚咪唑啉化合物,将咪唑啉引入高分子主链,具有良好的可加工性、高的热稳定性、可降解性。
[0037]
(2)本发明的超支化聚咪唑啉的制备方法,聚合反应具有良好的官能团耐受性,可以引入多种功能性基团。
[0038]
(3)本发明的超支化聚咪唑啉类化合物的制备方法,聚合反应实施过程工艺简单,反应原料易得,可直接购买或通过简单的反应制备;聚合反应条件温和,室温下就能聚合,节约能源;聚合效率高,反应1小时就能得到较高分子量的聚合物;聚合过程中无副产物生成,符合原子经济性。
[0039]
(4)本发明的超支化聚咪唑啉类化合物的制备方法,使用的催化剂氯化亚铜和三苯基膦,相比于其他金属催化剂体系,具有便宜易得的特点。
附图说明
[0040]
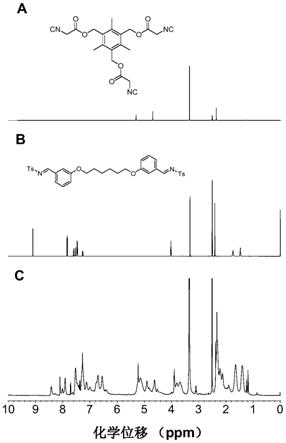
图1是聚合物p1a2a与其相应单体的红外吸收谱图。
[0041]
图2是聚合物p1a2a与其相应单体在dmso-d6中的核磁共振氢谱对比图。
[0042]
图3是聚合物p1a2a与其相应单体在dmso-d6中的核磁共振碳谱对比图。
[0043]
图4是聚合物p1a2b与其相应单体的红外吸收谱图。
[0044]
图5是聚合物p1a2b与其相应单体在dmso-d6中的核磁共振氢谱对比图。
[0045]
图6是聚合物p1a2b与其相应单体在dmso-d6中的核磁共振碳谱对比图。
[0046]
图7是聚合物p1b2a与其相应单体的红外吸收谱图。
[0047]
图8是聚合物p1b2a与其相应单体在dmso-d6中的核磁共振氢谱对比图。
[0048]
图9是聚合物p1b2a与其相应单体在dmso-d6中的核磁共振碳谱对比图。
[0049]
图10是聚合物p1b2b与其相应单体的红外吸收谱图。
[0050]
图11是聚合物p1b2b与其相应单体在dmso-d6中的核磁共振氢谱对比图。
[0051]
图12是聚合物p1b2b与其相应单体在dmso-d6中的核磁共振碳谱对比图。
[0052]
图13是聚合物p1a2b在酸性和碱性介质下的降解曲线。
具体实施方式
[0053]
下面结合实施例,对本发明作进一步地详细说明,但本发明的实施方式不限于此。
[0054]
实施例1
[0055]
(1)第一单体三元异腈基化合物1a的合成方法如下:在250ml的两口烧瓶中依次加入磁子、化合物1(2.513g,6.3mmol)和化合物2(2.436g,20mmol)。用橡胶塞塞住两口烧瓶侧口,中间口接通冷凝回流管,冷凝回流管上接氮气双排管。在严格按照schlenk line(双排管操作技术)重复三次抽真空通氮气后,用注射器通过侧口的橡胶塞加入约60ml dmf,于60℃下搅拌反应8小时。反应结束后,体系呈奶茶色混浊状,使其自然恢复至室温。用乙酸乙酯和饱和食盐水萃取三次,以去除体系中的dmf。往收集得到的有机相中加入无水硫酸镁干燥除水,抽滤,取滤液。往滤液中加入适量硅胶粉(100-200目),旋干做粉,将旋干得到的粉状混合物均匀加入到硅胶层析分离柱中,进一步通过硅胶柱层析法对产物进行分离提纯,以pe:ea的混合溶剂(2:1,v:v)为梯度淋洗剂,得到淡黄色粉末2-1a(1.81g,70%)。ft-ir(kbr disk),υ(cm-1
):2976,2150,1749, 1419,1380,1206,1001,919,821,720,574.1h nmr(500mhz,dmso-d6),δ (tms,ppm):5.30(s,6h),4.69(s,6h),2.35(s,9h).
13
c nmr(125mhz,dmso-d6), δ(tms,ppm):165.68,159.18,140.54,130.40,63.15,43.89,16.07.
[0056][0057]
(2)第二单体二元磺酰亚胺化合物2a的合成方法如下,
[0058]
中间体化合物5:在50ml的带支口的单口烧瓶中依次加入磁子、化合物3 (2.4g,21.2mmol)和碳酸钾(3.455g,2.5mmol),在支口处装上磨砂玻璃旋塞,并与氮气双排管相连,烧瓶口接通冷凝回流管,用橡胶塞塞住冷凝管关口。在严格按照schlenk line(双排管操作技术)重复三次抽真空通氮气后,用注射器往反应体系中加入1,6-二溴己烷4(1.5ml,10mmol)和7.5ml dmf,于110 ℃的条件下搅拌反应6小时。反应结束后,待反应体系自然恢复至室温,体系呈分层现象,下层为粘稠的固态物质,加入少许thf稀释溶解。将反应液倒入盛有300ml去离子水的烧杯中,充分搅拌,静置待其有固体析出后进行抽滤,滤饼用大量去离子水冲洗,滤饼干燥得粗产物。粗产物于正己烷中重结晶,抽滤干燥得纯化合物5,浅褐色晶体(2.452g,75%)。
[0059]
单体2a的合成:在50ml的两口烧瓶中依次加入磁子、化合物5(1.577g, 4.8mmol)和对甲苯磺酰胺(1.712g,10mmol)。用橡胶塞塞住两口烧瓶侧口,中间的瓶口放置一个防溅球用于收集反应副产物乙醇,防溅球上连接冷凝回流管,冷凝回流管上接氮气双排管。在严格按照schlenk line(双排管操作技术) 重复三次抽真空通氮气后,用注射器通过侧口的橡胶塞加入7.5ml硅酸四乙酯,于150℃下搅拌反应8小时。反应结束后,待反应体系自然恢复至室温,体系呈浅棕色混浊状态。将反应液倒入盛有150ml乙醚的烧杯中,充分搅拌,抽滤滤饼用大量乙醚冲洗,得纯净白色粉状化合物2a(2.28g,75%)。ft-ir(kbr disk), υ(cm
‑1):2942,1576,1446,1338,1260,1160,1088,1031,823,673,616,548.1h
ꢀꢀ
nmr(500mhz,dmso-d6),δ(tms,ppm):9.09(s,2h),7.88
–
7.78(m,4h),7.61
–
7.43(m,10h),7.26(j=8.2,2.6,0.8hz,2h),4.02(t,j=6.4hz,4h),2.40(s,6h), 1.74(j=10.6,4.7hz,4h),1.46(s,4h).
13
c nmr(125mhz,dmso-d6),δ(tms, ppm):171.86,159.34,145.11,135.18,133.89,130.33,128.06,124.20,122.71, 115.56,68.20,65.39,28.74,25.63,21.34,15.69.
[0060][0061]
(3)超支化聚咪唑啉p1a2a的合成与表征
[0062][0063]
在10ml带支口的schlenk管中依次加入磁子、单体1a(41.1mg,0.1mmol)、单体2a(63.3mg,0.1mmol)和三苯基膦(5.3mg,0.02mmol)。在重复三次抽真空通氮气后,在手套箱中称取1.0mg(0.01mmol)氯化亚铜,并用橡胶塞塞住 schlenk管管口。用注射器往反应体系中加入1ml二氯甲烷,待单体搅拌溶解,用10μl微量注射器往体系中加入2.8μl(0.02mmol)三乙胺,然后使整个体系于40℃下反应3小时。反应结束待其自然恢复至室温后,加入2ml二氯甲烷到反应液中稀释。将稀释好的反应液通过塞有棉花的玻璃滴管过滤装置逐滴加到150ml高速搅拌的正己烷溶剂中,得到白色絮状沉淀物。静置一段时间后用滤纸过滤,得到的沉淀物在40℃的真空干燥箱中干燥至恒重,得到目标聚合物p1a2a。
[0064]
本实施例制备的聚合物p1a2a为白色固体,产率:97%。凝胶渗透色谱(gpc) 结果显示:重均分子量(mw)为52000,分子量分布(pdi)为2.72。该超支化聚咪唑啉类化合物在室温下易溶于n,n-二甲基甲酰胺、二甲基亚砜等常见的有机溶剂,表明具有良好的可加工性。本实施例制备的聚合物5%,热失重温度为 243℃,表明该聚合物具有较好的热稳定性。ir(薄膜),ν(cm-1
):2942,2175,1742, 1607,1487,1451,1364,1264,1171,1093,1021,807.1h nmr(500mhz, dmso-d6),δ(tms,ppm):8.41,8.09,8.00,7.90,7.52,7.26,7.12,6.99,6.70,
6.54, 5.21,5.13,4.90,4.61,3.88,3.78,3.70,2.32,2.21,2.11,1.63,1.39.
13
c nmr(125 mhz,dmso-d6),δ(tms,ppm):170.07,169.51,162.10,159.34,145.08,142.23, 141.90,139.97,130.33,129.76,127.53,126.06,114.81,112.85,79.33,67.61,63.91, 62.05,46.21,29.16,25.88,21.48,15.91,9.12.
[0065]
本实施例制备的聚合物p1a2a(c)与其相应单体1a(a)和单体2a(b)的红外吸收谱图如图1所示,单体1a的异腈基团和c=o的伸缩振动分别出现在2150 和1746cm-1
处。单体2a的-c=n-ts的伸缩振动出现在1594cm-1
处。在聚合物 p1a2a的红外谱图中,2186cm-1
处的异腈基团特征峰又再次出现,这是因为在聚合反应过程中,异腈基团与磺酰亚胺基团的摩尔比为3:2,异腈基团过量,故聚合物中有未反应异腈基团;同样的,代表-c=n-ts的在1594cm-1
处的特征峰则转化为咪唑啉环上的-ch=n-的伸缩振动特征峰(1605cm-1
)。上述结果充分说明了异腈基和磺酰亚胺通过反应转化成了咪唑啉环。
[0066]
本实施例制备的聚合物(c)与其相应单体1a(a)、单体2a(b)在dmso-d6中的核磁共振氢谱对比图见图2,磺酰亚胺基团(-ch=n-ts)上氢(δ9.03)的峰在聚合物p1a2a的氢谱中已经完全消失。在聚合物p1a2a的氢谱中,新生成的咪唑啉环上的特征氢共振峰明显,δ7.88,4.84,4.58处的峰可归属于新生成的反式咪唑啉上三组氢的共振。核磁共振碳谱图3进一步验证了我们的结论。在聚合物p1a2a的碳谱中,新生成的咪唑啉环上的特征碳共振峰出现在δ150.49,78.98, 63.47三处。综上,可以认为通过三官能度异腈基乙酸酯与二元芳香磺酰亚胺的聚合成功得到了聚咪唑啉类超支化聚合物,且以反式结构为主。从图中可以确定该聚合物的核磁,精确证实了聚合物p1a2a的结构。
[0067]
实施例2~6
[0068]
实施例2~6考察了反应溶剂对此聚合反应的影响,聚合物单体的制备与实施例1相同。
[0069]
表1反应溶剂对单体1a和2a聚合的影响a[0070][0071]a在氮气氛围中,以cucl/pph3作为催化体系,在40℃下分别于不同溶剂中反应4小时;[1a]=[2a]=0.1m,[cucl/pph3]=1/2,[m]
cucl
=0.01m,[m]
tea
=0.02 m。
[0072]b通过gpc确定,以线性聚甲基丙烯酸甲酯为校正物,以dmf为流动相。
[0073]
考察了溶剂对聚合反应的影响,结果见表1。在使用四氢呋喃作为反应溶剂时,体
系出现凝胶现象,生成难溶物质,可能是四氢呋喃对产物的溶解性太差导致。在使用甲苯作为反应溶剂时,几乎得不到聚合物,这是由于单体在甲苯中的溶解性较差。当使用氯仿和n,n-二甲基甲酰胺作为溶剂时,反应都能以80%以上的收率得到分子量上万的聚合物,但以氯仿作溶剂时,其聚合物分子量分布相对较大;以n,n-二甲基甲酰胺作溶剂时,其收率相对较低。当使用dcm 作为反应溶剂时,可以得到最好的聚合结果,产率高达93%,产物重均分子量达到24200g/mol,多分散系数为2.09。综合考虑了产率、重均分子量、分子量分布等因素后,优先选择了二氯甲烷作为后续反应条件优化的溶剂。
[0074]
实施例7~10
[0075]
实施例7~10考察了不同的温度对反应条件的影响,聚合单体的制备与实施例1 相同,步骤(3)的反应条件和结果见表2。
[0076]
表2温度对单体1a和2a聚合的影响a[0077][0078]a在氮气氛围中,以二氯甲烷作为反应溶剂,以cucl/pph3作为催化体系,分别在不同温度下反应4小时;[1a]=[2a]=0.1m,[cucl/pph3]=1/2,[m]
cucl
=0.01m, [m]
tea
=0.02m。
[0079]b通过gpc确定,以线性聚甲基丙烯酸甲酯为校正物,以dmf为流动相。
[0080]
从表2中可以看出,无论是在冰浴条件下(0℃)、室温下(25℃)还是加热条件下,聚合反应都能发生,并且产率都很高(90%以上)。当反应温度低于 30℃时,聚合物的分子量随着反应温度的升高而增加,而在40℃时反应得到的聚合物分子量较在30℃时反应得到的聚合物分子量还要小,这可能是因为溶剂dcm的沸点在40℃左右,体系在40℃下进行反应会不稳定所导致。综上,优先选择30℃作为后续反应条件优化的温度。
[0081]
实施例11~17
[0082]
实施例11~17考察了不同的反应时间对反应条件的影响,聚合单体的制备与实施例1相同。
[0083]
表3反应时间对单体1a和2a聚合的影响a[0084][0085]a在氮气氛围中,以dcm作为反应溶剂,以cucl/pph3作为催化体系,在 30℃下分别反应不同的时间;[1a]=[2a]=0.1m,[cucl/pph3]=1/2,[m]
cucl
= 0.01m,[m]
tea
=0.02m。
[0086]bt=反应时间。
[0087]c通过gpc确定,以线性聚甲基丙烯酸甲酯为校正物,以dmf为流动相。
[0088]
从表3中可以看出,在延长反应时间的过程中,聚合反应被分成了两个阶段,前3小时为增长阶段,反应产率和聚合物分子量不断增长;3小时后,为降解阶段,分子量不断减少。这可能是三乙胺这种有机碱对聚合产物有降解作用,随着时间的延长,聚合产物的降解程度越高。综上,选择3小时作为后续反应条件优化的时间。
[0089]
实施例18~20
[0090]
实施例18~20考察了不同的单体浓度对反应条件的影响,聚合单体的制备与实施例1相同。
[0091]
表4单体浓度对单体1a和2a聚合的影响a[0092][0093]a在氮气氛围中,以二氯甲烷作为反应溶剂,以cucl/pph3作为催化体系,分别以不同的单体浓度在30℃下反应3小时;[1a]=[2a],[cucl/pph3]=1/2, [m]
cucl
=0.01m,[m]
tea
=0.02m。
[0094]b通过gpc确定,以线性聚甲基丙烯酸甲酯为校正物,以dmf为流动相。
[0095]
当反应体系的浓度较高时(0.2m),体系出现凝胶现象,生成难溶的凝胶物。当体系
浓度降低到0.05m时,聚合反应能够以高产率得到分子量上万的聚合物,但其分子量仅为浓度为0.1m时所得聚合物的一半。综上,选择0.1m作为后续反应优化的单体浓度。
[0096]
实施例21
[0097]
(1)第一单体三元异腈基化合物1a的合成方法同实施例1
[0098]
(2)第二单体二元磺酰亚胺类化合物2b的合成方法如下。
[0099]
中间体化合物8:在50ml的带支口的单口烧瓶中依次加入磁子、化合物6 (2.4g,21.2mmol)和碳酸钾(3.455g,2.5mmol),在支口处装上磨砂玻璃旋塞,并与氮气双排管相连,烧瓶口接通冷凝回流管,用橡胶塞塞住冷凝管关口。在严格按照schlenk line(双排管操作技术)重复三次抽真空通氮气后,用注射器往反应体系中加入1,6-二溴己烷7(1.5ml,10mmol)和7.5ml dmf,于110 ℃的条件下搅拌反应6小时。反应结束后,待反应体系自然恢复至室温,体系呈分层现象,下层为粘稠的固态物质,加入少许thf稀释溶解。将反应液倒入盛有300ml去离子水的烧杯中,充分搅拌,静置待其有固体析出后进行抽滤,滤饼用大量去离子水冲洗,滤饼干燥得粗产物。粗产物于正己烷中重结晶,抽滤干燥得纯化合物8,浅褐色晶体(2.452g,75%)。ft-ir(kbr disk),υ(cm-1
): 2943,2843,1590,1555,1515,1416,1309,1255,1148,1080,1015,764.1h nmr (500mhz,cdcl3),δ(tms,ppm):9.89,7.95,6.98,4.05,1.96,1.56.
13
c nmr(125 mhz,cdcl3),δ(tms,ppm):190.90,154.03,131.75,129.73,115.95,114.89,69.05, 29.13,25.79.
[0100]
单体2b的合成:在50ml的两口烧瓶中依次加入磁子、化合物8(1.577g, 4.8mmol)和对甲苯磺酰胺(1.712g,10mmol)。用橡胶塞塞住两口烧瓶侧口,中间的瓶口放置一个防溅球用于收集反应副产物乙醇,防溅球上连接冷凝回流管,冷凝回流管上接氮气双排管。在严格按照schlenk line(双排管操作技术) 重复三次抽真空通氮气后,用注射器通过侧口的橡胶塞加入7.5ml原硅酸四乙酯,于150℃下搅拌反应8小时。反应结束后,待反应体系自然恢复至室温,体系呈浅棕色混浊状态。将反应液倒入盛有150ml乙醚的烧杯中,充分搅拌,抽滤滤饼用大量乙醚冲洗,得纯净白色粉状化合物2b(2.22g,73%)。ft-ir(kbr
ꢀꢀ
disk),υ(cm-1
):2945,2846,1595,1560,1501,1422,1312,1253,1152,1077,1013, 876.1h nmr(500mhz,dmso-d6),δ(tms,ppm):9.03(s,2h),7.97(d,j=8.8hz, 4h),7.81(d,j=8.2hz,4h),7.44(d,j=8.1hz,4h),7.09(d,j=8.7hz,4h),4.09 (t,j=6.4hz,4h),2.40(s,6h),1.75(s,4h),1.46(s,4h).
13
c nmr(125mhz, dmso-d6),δ(tms,ppm):170.85,165.00,144.74,136.14,134.10,130.52,128.04, 125.19,115.80,68.47,28.74,25.43,21.62.
[0101]
[0102]
(3)超支化聚咪唑啉类化合物p1a2b的制备过程同实施例1
[0103][0104]
本实施例制备的聚合物为白色固体,产率:97%。凝胶渗透色谱(gpc)结果显示:重均分子量(mw)为208300g/mol,分子量分布(pdi)为8.89。该超支化聚咪唑啉类化合物在室温下易溶于n,n-二甲基甲酰胺、二甲基亚砜等常见的有机溶剂,表明具有良好的可加工性。本实施例制备的聚合物5%,热失重温度为228℃,表明该聚合物具有较好的热稳定性。ir(薄膜),ν(cm-1
):2937,2189, 1745,1685,1611,1517,1364,1245,1167,1092,1017,813.1h nmr(500mhz, dmso-d6),δ(tms,ppm):7.88,7.52,7.26,6.99,6.73,5.12,4.83,4.59,3.87,2.31, 2.23,2.10,1.67,1.43.
13
c nmr(125mhz,dmso-d6),δ(tms,ppm):168.56, 158.06,149.61,144.01,129.03,127.39,126.40,113.95,78.41,66.60,35.28,30.05, 28.12,24.84,20.60,14.70。
[0105]
本实施例制备的聚合物p1a2b(c)与其相应单体1a(a)和单体2b(b)的红外吸收谱图如图4所示,单体1a中,和c=o的伸缩振动分别出现在2150 和1746cm-1
处。同时,单体2b中的-c=n-ts的伸缩振动出现在1594cm-1
处。然而,在聚合物p1a2b的红外谱图中,2186cm-1
处的异腈基团特征峰又再次出现,这是因为在聚合反应过程中,异腈基团与磺酰亚胺基团的摩尔比为3:2,异腈基团过量,故聚合物中有未反应异腈基团;同样的,代表-c=n-ts的在1594cm-1
处的特征峰则转化为咪唑啉环上的-ch=n-的伸缩振动特征峰(1605cm-1
)。上述结果充分说明了异腈基和磺酰亚胺通过反应转化成了咪唑啉环。
[0106]
本实施例制备的聚合物(c)与其相应单体1a(a)、单体2b(b)在dmso-d6中的核磁共振氢谱和碳谱对比图见图5和图6,从图中可以确定该聚合物为聚咪唑啉类化合物。
[0107]
实施例22
[0108]
(1)第一单体三元异腈基化合物1b合成方法见下。
[0109]
中间体化合物11的合成:在250ml两口烧瓶中依次加入磁子和碳酸钾(6.9 g,50mmol),在两口烧瓶的侧口装上恒压滴液漏斗,中间的口接通冷凝回流管,冷凝回流管上接氮气双排管。在重复三次抽真空通氮气后,用注射器通过恒压滴液漏斗上的橡胶塞往反应烧瓶中加入80ml丙酮和1,6-二溴己烷(16ml,100 mmol),使其在80℃下加热搅拌。取一个100ml烧杯,准确称取化合物9(3.06 g,10mmol),加入适量丙酮将其完全溶解。待反应体系开始回流时,将配好的化合物9的丙酮溶液注射到恒压低液漏斗中,缓慢滴加,严格控制滴加速度,反应过夜。反应结束后,体系呈奶白色的混浊状态,使其自然恢复至室温,将体系中
的溶剂用旋转蒸发仪旋干,而后用二氯甲烷和饱和食盐水萃取三次。往收集得到的有机相中加入无水硫酸镁以干燥除水,抽滤,取滤液,往滤液中加入适量硅胶粉(100-200目),旋干做粉,将旋干得到的粉状混合物均匀加入到硅胶层析分离柱中,进一步通过硅胶柱层析法对产物进行分离提纯,以pe:dcm 的混合溶剂(30:1,v:v)为梯度淋洗剂,得到透明粘稠液体11(6.47g,81%)。
[0110]
单体1b的合成:在250ml的两口烧瓶中依次加入磁子、化合物9(5.01g, 6.3mmol)和化合物12(2.436g,20mmol)。用橡胶塞塞住两口烧瓶侧口,中间口接通冷凝回流管,冷凝回流管上接氮气双排管。在严格按照schlenk line(双排管操作技术)重复三次抽真空通氮气后,用注射器通过侧口的橡胶塞加入约 60ml dmf,于60℃下搅拌反应8小时。反应结束后,体系呈奶茶色混浊状,使其自然恢复至室温。用ea和饱和食盐水萃取三次,以去除体系中的dmf。往收集得到的有机相中加入无水硫酸镁以干燥除水,抽滤,取滤液。往滤液中加入适量硅胶粉(100-200目),旋干做粉,将旋干得到的粉状混合物均匀加入到硅胶层析分离柱中,进一步通过硅胶柱层析法对产物进行分离提纯,以pe:ea 的混合溶剂(2:1,v:v)为梯度淋洗剂,得到无色透明粘稠液体1b(3.38g,70%)。 ft-ir(kbr disk),υ(cm-1
):2953,2150,1753,1685,1602,1503,1472,1394,1352, 1244,1010,876.1h nmr(500mhz,dmso-d6),δ(tms,ppm):6.90(d,j=8.9hz, 6h),6.80(d,j=9.0hz,6h),4.14(t,j=6.6hz,6h),3.91(t,j=6.4hz,6h),2.01 (s,3h),1.75
–
1.65(m,6h),1.65
–
1.57(m,6h),1.46
–
1.31(m,12h).
13
c nmr (125mhz,dmso-d6),δ(tms,ppm):165.64,159.25,157.15,141.81,129.65, 114.04,67.64,66.12,50.51,44.06,28.93,28.25,25.42.
[0111][0112]
(2)第二单体二元磺酰亚胺类化合物2a的合成方法同实施例1。
[0113]
(3)超支化聚咪唑啉类化合物p1b2a的制备过程同实施例1。
[0114][0115]
本实施例制备的聚合物p1b2a为白色粉末,产率:92%。凝胶渗透色谱(gpc) 结果显示:重均分子量(mw)为138200g/mol,分子量分布(pdi)为5.04。该超支化聚咪唑啉类化合物在室温下易溶于n,n-二甲基甲酰胺、二甲基亚砜等常见的有机溶剂,表明具有良好的可加工性。本实施例制备的聚合物5%热失重温度为244℃,表明具有非常高的热稳定性。ir(薄膜),ν(cm-1
):2942,2175,1741, 1607,1508,1364,1244,1176,1109,1015,818,667.1h nmr(500mhz,dmso-d6), δ(tms,ppm):8.40,8.09,8.01,7.91,7.56,7.49,7.39,7.26,7.11,7.01,6.85,6.73, 6.61,6.43,5.18,4.85,4.56,3.82,2.30,1.96,1.61,1.37,0.96.
13
c nmr(125 mhz,dmso-d6),δ(tms,ppm):168.38,167.15,158.26,157.60,155.91,144.01, 140.59,139.50,133.54,132.63,129.26,128.36,128.17,126.45,125.01,112.75, 98.92,66.49,49.54,30.35,27.98,24.49,21.37,20.40,13.41。
[0116]
本实施例制备的聚合物p1b2a(c)与其相应单体1b(a)和单体2a(b)的红外吸收谱图如图7所示,单体1b中,异腈的伸缩振动分别出现在2150cm-1
处。同时,单体2a中的-c=n-ts的伸缩振动出现在1594cm-1
处。然而,在聚合物 p1b2a的红外谱图中,2186cm-1
处的异腈基团特征峰又再次出现,这是因为在聚合反应过程中,异腈基团与磺酰亚胺基团的摩尔比为3:2,异腈基团过量,故聚合物中有未反应异腈基团;同样的,代表-c=n-ts的在1594cm-1
处的特征峰则转化为咪唑啉环上的-ch=n-的伸缩振动特征峰(1605cm-1
)。上述结果充分说明了异腈基和磺酰亚胺通过反应转化成了咪唑啉环。
[0117]
本实施例制备的聚合物(c)与其相应单体1b(a)、单体2a(b)在dmso-d6中的核磁共振氢谱和碳谱对比图见图8和图9,从图中可以确定该聚合物为聚咪唑啉类化合物。
[0118]
实施例23
[0119]
(1)第一单体三元异腈基化合物1b合成方法同实施例22。
[0120]
(2)第二单体二元磺酰亚胺化合物2b的合成方法同实施例21。
[0121]
(3)超支化聚咪唑啉类化合物p1b2b的制备过程同实施例1
[0122][0123]
本实施例制备的聚合物为白色粉末,产率:97%。凝胶渗透色谱(gpc)结果显示:重均分子量(mw)为62300,分子量分布(pdi)为5.33。该聚咪唑啉类化合物在室温下易溶于n,n-二甲基甲酰胺、二甲基亚砜等常见的有机溶剂,表明具有良好的可加工性。本实施例制备的聚合物5%热失重温度为242℃,表明具有高的热稳定性。ir(薄膜),ν(cm-1
):2956,2174,1743,1617,1509,1472,1368, 1250,1172,1104,1016,819.1h nmr(500mhz,dmso-d6),δ(tms,ppm):8.09, 7.48,7.26,7.02,6.86,6.73,6.62,5.13,4.81,3.84,3.49,3.10,2.31,1.96,1.61,1.35, 0.92.
13
c nmr(125mhz,dmso-d6),δ(tms,ppm):162.08,159.20,157.04,141.64, 130.33,129.63,127.60,126.05,114.97,114.01,67.60,65.51,64.78,50.53,46.24, 31.43,28.98,25.52,22.53,21.47,14.35,9.08.
[0124]
本实施例制备的聚合物p1b2b(c)与其相应单体1b(a)和单体2b(b)的红外吸收谱图如图10所示,单体1b中,异腈的伸缩振动分别出现在2150cm-1
处。同时,单体2b中的-c=n-ts的伸缩振动出现在1594cm-1
处。然而,在聚合物 p1b2b的红外谱图中,2186cm-1
处的异腈基团特征峰又再次出现,这是因为在聚合反应过程中,异腈基团与磺酰亚胺基团的摩尔比为3:2,异腈基团过量,故聚合物中有未反应异腈基团;同样的,代表-c=n-ts的在1594cm-1
处的特征峰则转化为咪唑啉环上的-ch=n-的伸缩振动特征峰(1605cm-1
)。上述结果充分说明了异腈基和磺酰亚胺通过反应转化成了咪唑啉环。
[0125]
本实施例制备的聚合物(c)与其相应单体1b(a)、单体2b(b)在dmso-d6中的核磁共振氢谱和碳谱对比图见图11和图12,从图中可以确定该聚合物为聚咪唑啉类化合物。
[0126]
实施例24
[0127]
多官能度异腈基乙酸酯与二元芳香磺酰亚胺聚合得到的超支化聚合物中含有亲水的酯基,酯基在水中或生物介质中可发生水解,使聚合物的分子链发生断裂,达到降解的目的。实验中考察了降解介质对聚合物降解规律的影响,具体实验步骤如下:
[0128]
在50ml茄形瓶中依次加入磁子、10mg超支化聚合物p1a2b(mw=208400 g/mol)、2ml超干dmf、100μl浓hcl(12mol/l)、100μl h2o,用磨口玻璃塞塞住瓶口于室温下搅拌,每隔一小时取0.2ml反应液转移至装有3ml乙醚的塑料离心管中使其沉淀,离心,干燥,得降解不同时间后的聚合物,用凝胶渗透色谱仪(gpc)测定其分子量。
[0129]
对于使用有机碱三乙胺(tea)作为降解介质的实验步骤同上类似,在50ml 茄形瓶中一次加入磁子、10mg超支化聚合物p1a2b(mw=208400g/mol)、2ml 超干dmf、200μl超干三乙胺,用磨口玻璃塞塞住瓶口于室温下搅拌,每隔 30分钟取0.2ml反应液转移至装有3ml乙
醚的塑料离心管中使其沉淀,离心,干燥,得降解不同时间后的聚合物,用凝胶渗透色谱仪(gpc)测定其分子量。
[0130]
超支化聚合物p1a2b的分子量随降解时间的变化如图13所示。可以看出,碱性条件更有利于聚合物的降解,仅0.5小时就使其分子量下降为原来的十分之一;而酸性条件下,10小时后其分子量才下降为原来的三分之一。上述结果表明,该聚合物可在酸性条件和碱性条件下发生降解,但在碱性环境下降解更迅速更完全,具有环境友好性。
[0131]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受所述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。