一种
α-熊果苷的提取方法和应用
技术领域
1.本发明属于生物化工技术领域,具体涉及一种α-熊果苷的提取方法和应用。
背景技术:
2.熊果苷,又名熊果素,其作为酪氨酸酶的抑制剂,能阻断多巴及多巴醌的合成,从而有效抑制黑色素的生成,发挥美白作用,且对皮肤没有刺激性,毒副作用小。熊果苷具有显著抑制酪氨酸酶活性的作用,是一种新兴的无刺激、无过敏和配伍性强的天然美白活性物质,主要应用于化妆品中。目前,化妆品中常用的两种熊果苷分别是α-熊果苷与β-熊果苷,其中α-熊果苷的美白活性是β-熊果苷的十倍。
3.氢醌作为酶法生产α-熊果苷的主要原料,在α-熊果苷产品中有残留。氢醌在世界卫生组织国际癌症研究机构公布的致癌物清单的3类致癌物中,同时《q/jsb 001-2016化妆品用原料α-熊果苷质量标准》对氢醌的含量进行了规定,限定在10ppm以内,另外,当α-熊果苷应用于质量要求更高的食品级、医药级原料时,要求会更高。α-熊果苷中残留的氢醌如若不能有效除去,会直接影响产品质量甚至直接导致产品不合格。目前常见的α-熊果苷提纯方法有溶剂萃取法、吸附解析法等,这些方法虽技术成熟,但会用到大量的有机溶剂,或需要大量酸碱再生树酯,产生三废较多、污染环境,增加处理成本,且工艺成本高,不适宜工业化生产。
技术实现要素:
4.本发明的目的在于提供一种α-熊果苷的提取方法,包括下述步骤:
5.(1)将α-熊果苷转化液调ph至酸性后,进行连续色谱分离,收集α-熊果苷料液;
6.(2)将α-熊果苷料液脱色,脱色清液经浓缩、降温、析晶、分离,得α-熊果苷粗品;
7.(3)将α-熊果苷粗品经重结晶、烘干,得α-熊果苷成品。
8.本发明的优选技术方案,步骤(1)中,α-熊果苷转化液调ph为2-6,优选为3-5,更优选为4-5。
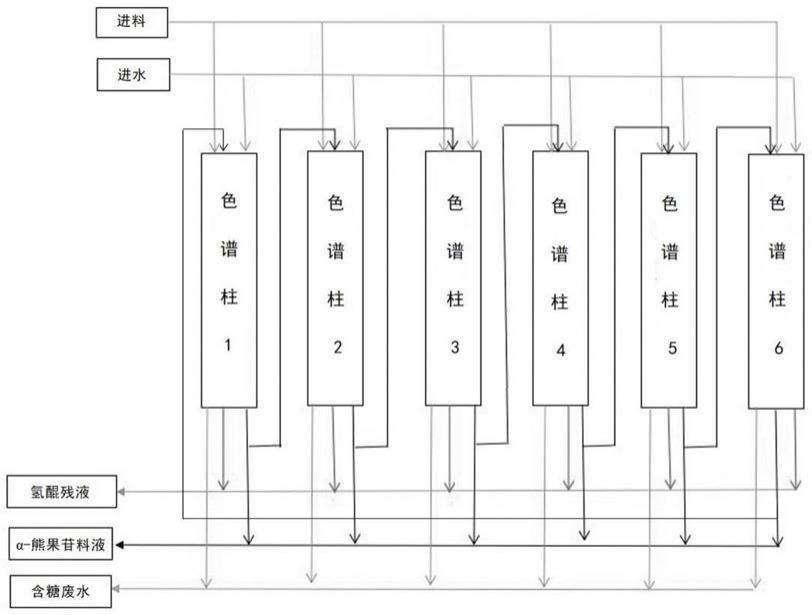
9.本发明的优选技术方案,α-熊果苷转化液在调ph前可先过滤;优选地,α-熊果苷转化液先通过陶瓷膜,再通过卷式膜。
10.本发明的优选技术方案,所述连续色谱分离中所用的色谱柱填料为弱酸性阳离子交换树脂,优选为d001、d201、d301、d113中的任一种。
11.本发明的优选技术方案,所述连续色谱分离中所用的色谱柱串联连接。
12.本发明的优选技术方案,所述连续色谱分离中所用的色谱柱至少为2根,优选为2-6根色谱柱串联,更优选为3-4根色谱柱串联,再优选为4根色谱柱串联。
13.本发明的优选技术方案,所述连续色谱分离为:
14.将用于连续色谱分离的色谱柱串联连接,组成色谱柱组;
15.从色谱柱组的第1根色谱柱开始按照设定流速进料,从第1根色谱柱开始,依次在各根色谱柱开始出料后先收集设定量的含糖废水,然后控制料液继续传动至串联连接的后
续色谱柱中;
16.在设定进料量进料完成后,开始从第1根色谱柱进洗脱液,并继续依次在后续色谱柱出口收集设定量的含糖废水;
17.在色谱柱组的所有色谱柱均收集完成设定量的含糖废水后,将色谱柱组的最后1根色谱柱切换收集设定量的α-熊果苷料液。
18.本发明的优选技术方案,进料量不大于色谱柱组各色谱柱体积之和,优选为小于色谱柱组各色谱柱体积之和。
19.本发明的优选技术方案,进料流速为0.5-2bv/h,优选为1-2bv/h。
20.本发明的优选技术方案,所述色谱柱组中各色谱柱的含糖废水收集量为0.3-0.6倍柱体积,优选为0.4-0.5倍柱体积。
21.本发明的优选技术方案,所述洗脱液体积不少于连续色谱系统各色谱柱体积之和,优选为等于连续色谱系统各色谱柱体积之和。
22.本发明的优选技术方案,所述洗脱液为纯水。
23.本发明的优选技术方案,所述连续色谱系统所含总色谱柱数量多出连续色谱分离中所用的色谱柱至少1根,优选为多出2根。
24.本发明的优选技术方案,在α-熊果苷料液收集完成后,将首轮连续色谱分离中,进洗脱液前已出料的色谱柱与首轮连续色谱分离中未使用的色谱柱重新串联组成色谱柱组,用于下一轮连续色谱分离;将进洗脱液后才出料的色谱柱加洗脱液顶洗待用。
25.本发明的优选技术方案,所述顶洗的洗脱液用量不少于待顶洗的总色谱柱体积,优选为大于待顶洗的总色谱柱体积。
26.本发明的优选技术方案,步骤(2)中,将经连续色谱分离获得的α-熊果苷料液通过活性炭脱色;优选地,通过0.5-1.0%活性炭脱色,脱色温度为30-80℃,脱色时间为0.5-2h;更优选地,通过0.5%活性炭脱色,脱色温度为50-60℃,脱色时间为1.0-1.5小时。
27.本发明的优选技术方案,脱色后的α-熊果苷料液经真空浓缩至α-熊果苷含量500-600g/l。
28.本发明的优选技术方案,所述真空浓缩的真空度为-0.07-0.1mpa,优选为-0.08-0.1mpa,更优选为-0.09-0.1mpa;浓缩温度为40-80℃,优选为50-75℃,更优选为55-60℃。
29.本发明的优选技术方案,所述降温的温度为10-30℃,维持1-4h;优选地,所述降温的温度为10-20℃,维持2-4h;更优选地,所述降温的温度为10-15℃,维持2-3h。
30.本发明的优选技术方案,步骤(3)中,所述重结晶包括以下步骤:
31.a、将α-熊果苷粗品用纯水加热溶解;
32.b、将上述溶解体系降温,析晶。
33.本发明的优选技术方案,所述溶解体系中α-熊果苷粗品含量为500-600g/l。
34.本发明的优选技术方案,步骤a中,加热温度为40-80℃,优选为50-70℃,更优选为55-60℃。
35.本发明的优选技术方案,步骤b中,降温温度为10-30℃,优选为10-20℃,更优选为10-15℃。
36.本发明的优选技术方案,所述烘干为50-80℃、-0.07-0.1mpa下真空干燥4-12h,优选为50-70℃、-0.08-0.1mpa下真空干燥5-10h,更优选为60-65℃、-0.09-0.1mpa下真空干
燥6-8h。
37.本发明的另一目的在于提供一种α-熊果苷,所述α-熊果苷纯度≥99.5%,优选≥99.6%,更优选≥99.7%,再优选≥99.8%,还优选≥99.9%。
38.本发明的优选技术方案,所述α-熊果苷中氢醌含量<10ppm,优选<8ppm,更优选<6ppm,再优选<4ppm,还优选<2ppm,最优选为0。
39.本发明的另一目的在于提供本发明所述α-熊果苷在制备化妆品中的应用。
40.本发明的优选技术方案,所述化妆品包括美白护肤品、淡斑护肤品、抗炎护肤品、防晒护肤品。
41.本发明的另一目的还在于提供本发明所述α-熊果苷在制备消炎药物中的应用。
42.本发明的另一目的还在于提供本发明所述α-熊果苷在制备抑制黑色素生成的药物中的应用。
43.除非另有说明,本发明涉及液体与液体之间的百分比时,所述的百分比为体积/体积百分比;本发明涉及液体与固体之间的百分比时,所述百分比为体积/重量百分比;本发明涉及固体与液体之间的百分比时,所述百分比为重量/体积百分比;其余为重量/重量百分比。
44.与现有技术相比,本发明具有下述有益技术效果:
45.1、本发明提供了一种α-熊果苷的提取方法,该提取方法将α-熊果苷转化液调ph后进行连续色谱分离,实现含糖废水、氢醌残液和α-熊果苷溶液的有效分离,所收集的α-熊果苷溶液纯度很高,氢琨含量低,满足化妆品等使用需求。另外,将收集的α-熊果苷溶液进行脱色、浓缩、降温结晶、离心、重结晶、烘干获得α-熊果苷成品,α-熊果苷纯度可高达99.99%,同时将产品中氢醌含量降至1ppm以下,甚至为0,α-熊果苷成品满足作为化妆品或药品添加剂的安全性能要求,可用于制备抑制黑色素生成的药物、消炎药物和/或化妆品。
46.2、本发明的连续色谱分离方法通过对进料量、进料速度、分离工作的色谱柱连接方式及串联工作的色谱柱数量、含糖废液排出量等进行优化设计,获得可高收率提取α-熊果苷的方法,制得的α-熊果苷纯度高、氢醌含量低甚至完全去除,满足化妆品原料要求,适宜工业化生产。
47.3、本发明通过连续色谱系统的快速重组可实现对α-熊果苷待提取液进行连续分离工作,节约工作时间,提高工作效率,降低生产成本。
48.4、本发明采用纯水作为洗脱剂,可避免产生有毒三废,环境友好。
附图说明
49.图1实施例1的连续色谱系统分离提取α-熊果苷的流程图。
具体实施方式
50.下面结合实施例对本发明的技术方案作进一步详细的说明。以下实施例仅用于说明本发明而不用于限制本发明的范围。下述实施例中所使用的实验方法,如无特殊说明,均为常规方法;所用的试剂和材料,如无特殊说明,均可从商业途径获得。
51.制备例1α-熊果苷透过液的制备
52.1、制备α-熊果苷转化液
53.(1)制备含蔗糖磷酸化酶菌液:枯草芽孢杆菌经种子培养和发酵培养后,收集含蔗糖磷酸化酶菌液;
54.(2)制备α-熊果苷转化液:将3400g蔗糖溶解在7l纯水中,加入132g氢醌溶解完全后加纯水定容至10l,调节反应溶液的ph为6.8,加入100ml含蔗糖磷酸化酶菌液并于30℃下转化反应3h,获得α-熊果苷转化液;
55.经高效液相色谱法(hplc)检测,步骤(2)制备的α-熊果苷转化液中α-熊果苷含量为29.8g/l,氢醌含量为0.85g/l;
56.2、将步骤1所得α-熊果苷转化液过陶瓷膜,收集过滤液;然后将过滤液过2000分子量超滤膜,收集透过液;透过液中α-熊果苷含量为29.76g/l,氢醌含量为0.85g/l。
57.实施例1
58.本发明的一种α-熊果苷的提取方法,采用连续色谱系统进行分离提取,包括下述步骤:
59.1、取2000ml制备例1所得的透过液调节ph为5,作为待提取液;
60.其中,所用连续色谱系统共有6根色谱柱,各色谱柱的体积均为500ml,所用色谱柱填料为弱酸性阳离子交换树脂d001;
61.2、将上述调ph后的待提取液通过连续色谱系统过色谱柱分离,具体包括下述步骤:每次4根色谱柱串联组成色谱柱组(如图1所示,色谱柱1、色谱柱2、色谱柱3、色谱柱4)进行连续分离工作;色谱柱组工作时,从色谱柱1开始进料,进4倍柱体积料液,进料流速2bv/h,色谱柱1开始出料后先收集0.5倍柱体积的含糖废水,然后色谱柱1停止收集;色谱柱2开始出料后,收集0.5倍柱体积的含糖废水,然后色谱柱2停止收集;色谱柱3开始出料后,开始从色谱柱1进纯水,纯水量为6倍柱体积,依次在色谱柱3和色谱柱4出口各收集0.5倍柱体积的含糖废水,然后将色谱柱4切换收集4倍柱体积的α-熊果苷料液,然后选取色谱柱1、色谱柱2、色谱柱5、色谱柱6重新串联组成色谱柱组工作(可选择柱6连接柱1,重组后由柱5开始进料,色谱柱串联顺序为5612),色谱柱3和色谱柱4加纯水顶洗3倍柱体积待用,废水为氢醌残液;
62.经连续色谱分离的α-熊果苷收率为90.4%,α-熊果苷料液中氢醌含量为0.00146g/l,α-熊果苷料液含糖量为0.05g/l;
63.3、将收集的α-熊果苷料液通过0.5%活性炭于60℃下脱色60min后,60℃、-0.09mpa下真空浓缩至α-熊果苷含量为500-600g/l,然后降温至10℃,维持2h至大量晶体析出,晶体离心,得60.6gα-熊果苷粗品。
64.将60.6gα-熊果苷粗品与80ml纯水混合,加热至60℃溶解;将上述溶解体系降温至10℃,维持5h以上进行,获得α-熊果苷晶体;将α-熊果苷晶体离心,然后在60℃、-0.1mpa下真空干燥6h,得29.25gα-熊果苷成品,α-熊果苷纯度为99.91%,氢醌为0.43ppm。
65.实施例2
66.本发明的一种α-熊果苷的提取方法,采用同实施例1的连续色谱系统,包括下述步骤:
67.1、取2000ml制备例1所得待提取液调节ph为5,作为连续色谱分离的待分离原料;
68.2、将上述待分离原料通过连续色谱系统过色谱柱分离,具体包括下述步骤:每次4根色谱柱串联组成色谱柱组(如图1所示,色谱柱1、色谱柱2、色谱柱3、色谱柱4)进行连续分
离工作;色谱柱组工作时,从色谱柱1开始进料,进4倍柱体积料液,进料流速1bv/h,色谱柱1开始出料后先收集0.5倍柱体积的含糖废水,然后色谱柱1停止收集;色谱柱2开始出料后,收集0.5倍柱体积的含糖废水,然后色谱柱2停止收集;色谱柱3开始出料后,开始从色谱柱1进纯水,纯水量为6倍柱体积,依次在色谱柱3和色谱柱4出口各收集0.5倍柱体积的含糖废水,然后将色谱柱4切换收集4倍柱体积的α-熊果苷料液,然后选取色谱柱1、色谱柱2、色谱柱5、色谱柱6重新串联组成色谱柱组工作,色谱柱3和色谱柱4进行加3倍柱体积的纯水顶洗待用,废水为氢醌残液;
69.经连续色谱分离的α-熊果苷收率为97.2%,α-熊果苷料液中氢醌含量为0.00023g/l,含糖量为0;
70.3、将收集的α-熊果苷料液经0.5%活性炭于60℃下脱色60min后,60℃、-0.1mpa下真空浓缩至α-熊果苷含量为500-600g/l,然后降温至10℃,维持2h,至大量晶体析出,晶体离心,得66.75gα-熊果苷粗品;
71.将66.75g粗品α-熊果苷与80ml纯水混合,加热至60℃溶解;将上述溶解体系降温至10℃,维持5h以上进行,获得α-熊果苷晶体;将α-熊果苷晶体离心,然后在60℃、-0.1mpa下真空干燥6h,得31.15gα-熊果苷成品,α-熊果苷纯度为99.94%,氢醌为0.12ppm。
72.实施例3
73.本发明的一种α-熊果苷的提取方法,采用同实施例1的连续色谱系统,包括下述步骤:
74.1、取1500ml制备例1所得待提取液调节ph为5,作为连续色谱分离的待分离原料;
75.2、将上述待分离原料通过连续色谱系统过色谱柱分离,具体包括下述步骤:每次4根色谱柱串联组成色谱柱组(如图1所示,色谱柱1、色谱柱2、色谱柱3、色谱柱4)进行连续分离工作;色谱柱组工作时,从色谱柱1开始进料,进3倍柱体积料液,进料流速1bv/h,色谱柱1开始出料后先收集0.5倍柱体积的含糖废水,然后色谱柱1停止收集;色谱柱2开始出料后,收集0.5倍柱体积的含糖废水,然后色谱柱2停止收集;开始从色谱柱1进纯水,纯水量为6倍柱体积,依次在色谱柱3和色谱柱4出口各收集0.5倍柱体积的含糖废水,然后将色谱柱4切换收集4倍柱体积的α-熊果苷料液,然后选取色谱柱1、色谱柱2、色谱柱5、色谱柱6重新串联组成色谱柱组工作,色谱柱3和色谱柱4进行加3倍柱体积的纯水顶洗待用,废水为氢醌残液;
76.经连续色谱分离的α-熊果苷收率为99.3%,α-熊果苷料液中氢醌含量为0,含糖量为0;
77.3、将收集的α-熊果苷料液经0.5%活性炭于60℃下脱色60min后,55℃、-0.1mpa下真空浓缩至α-熊果苷含量为500-600g/l,然后降温至10℃,维持4h,至大量晶体析出,晶体离心,得49.6gα-熊果苷粗品;
78.将49.6g粗品α-熊果苷与70ml纯水混合,加热至60℃溶解;将上述溶解体系降温至10℃,维持5h以上进行,获得α-熊果苷晶体;将α-熊果苷晶体离心,然后在60℃、-0.1mpa下真空干燥6h,得24.8gα-熊果苷成品,α-熊果苷纯度为99.99%,氢醌含量为0。
79.实施例4
80.本实施例与实施例3的区别在于,色谱柱组的最后一个色谱柱(如一轮分离的色谱柱4)切换收集3倍柱体积的α-熊果苷料液,α-熊果苷收率为74.45%,α-熊果苷料液中氢醌
含量为0,含糖量为0。
81.将收集的α-熊果苷料液经0.5%活性炭脱色、浓缩、降温、析晶、晶体离心,重结晶、干燥(实验方法和条件同实施例3),得到19.23gα-熊果苷成品,α-熊果苷纯度为99.99%,氢醌含量为0ppm。
82.实施例5
83.本实施例与实施例3的区别在于,色谱柱组的最后一个色谱柱(如一轮分离的色谱柱4)切换收集5倍柱体积的α-熊果苷料液,α-熊果苷收率为99.35%,α-熊果苷料液中氢醌含量为0.025g/l,含糖量为0。
84.将收集的α-熊果苷料液经0.5%活性炭脱色、浓缩、降温、析晶、晶体离心,重结晶、干燥(实验方法和条件同实施例3),得到25.06gα-熊果苷成品,α-熊果苷纯度为99.89%,氢醌含量为13ppm。
85.实施例6
86.本实施例与实施例3的区别在于,步骤2中,色谱柱组工作时,从色谱柱1开始进料,进3倍柱体积料液,进料流速1bv/h,色谱柱1开始出料后先收集0.4倍柱体积的含糖废水,然后色谱柱1停止收集;色谱柱2开始出料后,收集0.4倍柱体积的含糖废水,然后色谱柱2停止收集;继续进料至色谱柱3中,待3倍柱体积料液进料完成后,开始从色谱柱1进纯水,纯水量为6倍柱体积,依次在色谱柱3和色谱柱4出口各收集0.4倍柱体积的含糖废水,然后将色谱柱4切换收集4倍柱体积的α-熊果苷料液,然后选取色谱柱1、色谱柱2、色谱柱5、色谱柱6重新串联组成色谱柱组工作,色谱柱3和色谱柱4进行加3倍柱体积的纯水顶洗待用,废水为氢醌残液;
87.经连续色谱分离的α-熊果苷收率为99.38%,α-熊果苷料液中氢醌含量为0,含糖量为0.26g/l。
88.将收集的α-熊果苷料液经0.5%活性炭脱色、浓缩、降温、析晶、晶体离心,重结晶、干燥(实验方法和条件同实施例3),得到25.31gα-熊果苷成品,其中α-熊果苷纯度为99.81%,氢醌含量为0ppm,含糖量为0.09%。
89.以上仅是本发明的优选实施方式,本发明的保护范围并不仅局限于上述实施例,凡属于本发明思路下的技术方案均属于本发明的保护范围。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理前提下的若干改进和润饰,应视为本发明的保护范围。