1.本发明涉及微生物鉴定领域,具体而言,涉及一种通过测序获取微生物物种及相关信息的方法、装置、计算机可读存储介质和电子设备。
背景技术:
2.微生物划分为以下8大类:细菌、病毒、真菌、放线菌、立克次氏体、支原体、衣原体、螺旋体。二代测序(ngs)(mngs)技术是鉴定样本中存在的微生物种属类别的有效方法。
3.在微生物鉴定方面,ngs的应用主要有两种方法:
4.一种是使用宏基因组策略。检测样本中所有的核酸序列,通过将检测到的序列与微生物基因组序列数据库比对,识别样本中存在的微生物。
5.另外一种是靶向测序策略。特异性的捕获或者富集样本中的某些特征性序列,然后再进行序列检测,并将获得的序列信息与相应的微生物特征性序列数据库比对,从而鉴定样本中存在的微生物。原核生物的rrna有3种类型:23s、16s、5srrna。编码16s rrna的基因具有良好的进化保守性,适宜分析的长度(约为1540bp),以及与进化距离相匹配的良好变异性,所以成为细菌分子鉴定的标准标识序列。16s rrna基因不仅适用于细菌,也适用于支原体、衣原体、立克次氏体、螺旋体等原核生物的分类,是目前针对原核生物分类时,得到广泛接受、数据库最齐全的特征性序列。16s rrna的序列包含9或10个高变区(variable region)和10或11个保守区(conserved region)。保守区序列反映了生物物种间的亲缘关系,而高变区序列则体现物种间的差异。ngs靶向测序策略是针对16s rrna基因的高变区序列。pcr扩增100-几百bp的序列被用于ngs测序,获得的序列信息经与16s rrna基因序列数据库比对后,可鉴定样本中存在的微生物。
6.但是,当宏基因组二代测序(mngs)技术应用于微生物鉴定、特别是临床病原微生物鉴定时,mngs对样本中的核酸不加区分的全部测序。由于样本中存在大量非微生物的宿主核酸如大量来自人体细胞的人类基因组核酸,而一个人体细胞的核酸量大约是微生物的1,000-100,000倍,且微生物中具有种属特异性的序列仅有自身基因组的大约1/100,加之临床样本中病原微生物的数量相比宿主细胞的数量而言非常稀少,因而,被检测的样本核酸中仅有1/1,000,000-100,000,000的量来自病原微生物,这就导致绝大多数的测序数据都与微生物的鉴定无关,属于无效数据。测序数据的浪费,一方面导致检测成本高昂,另一方面由于有效数据过少,降低了检测的灵敏度与可靠性。
7.而基于16s/18s/its扩增子的ngs技术则读长有限,取决于不同的测序平台,测序片段的长度介于50~400bp之间。16s rrna基因的长度约1500bp。为了获得该基因全长的序列信息,必须将该基因核酸分子打断成适合ngs测序的短片段,并在测序完成后根据不同短片段相互间在末端序列上的重叠,将短片段按基因序列的顺序叠放后组装出16s rrna基因序列的全长。然而,由于核糖体基因序列在物种进化过程中高度保守,进化关系相近物种(如相同属(genus)内的物种)的序列相似度很高。因此,在含有一个以上物种的临床样本
中,由于分属不同物种的短片段间的序列相似性太高,使得从短片段正确地组装出各物种的全长16s rrna基因序列而不出现跨物种混合体(chimera)是难以实现的。
8.为避免上述困难,当前流行的扩增子二代测序(ngs)技术对16s rrna基因的高变区进行靶向扩增,而后对扩增子(amplicon)进行ngs测序。由于16s rrna的序列所包含的9或10个高变区(variable region)的核苷酸序列体现了物种间的差异,因此,通过对高变区的ngs测序、并将测序得到的序列信息与16srrna基因高变区序列数据库比对,可以获得对部分微生物在“种”(species)的水平上的鉴别能力。
9.但是,单个或若干个高变区所携带的核苷酸序列多样性并不足以用来区分所有的原核微生物。johnson,j.s.et al.(2019)的研究表明,只有全长的16s rrna基因核苷酸序列才包含足够的信息以在“种”的水平上区分所有的原核微生物。因此,当前的16s/18s/its扩增子二代测序(ngs)技术无法达到在“种”(species)的水平上检测临床样本中微生物的鉴别能力。
10.综上所述,上述方案应用于临床微生物检测时均存在各自无法克服的缺陷和不足。
技术实现要素:
11.本发明涉及一种通过测序获取微生物物种及相关信息的方法,包括:
12.i)获取测序数据,所述测序数据由靶向富集微生物特征核酸序列后经下一代测序技术对扩增产物进行测序得到;
13.ii)将所述测序数据与特征序列数据库进行比对分析以鉴定待检测样本中的微生物组成;
14.其中所述特征序列数据库预先根据含有所述特征序列的参考序列的相似性做聚类处理,得到一层或多层级的cluster,每个层级cluster中有至少一个子seed,最底层的cluster中有一或若干作为参考序列的seed;
15.所述比对分析包括:
16.a)去除所述测序数据中的低质量和含有测序接头序列的reads;
17.b)将reads序列与所述特征序列数据库的seeds序列进行比对,将因pcr扩增而产生的完全重复reads去除,并对事件“有reads序列比对上”与seeds序列做统计学独立性检验,选出与事件“有reads序列比对上”相关的seeds序列,得到初级筛选seed序列;
18.c)将read序列与所述初级筛选seed序列进行比对,其中所述初级筛选seed序列之间不竞争reads,计算每个seed序列的reads覆盖情况,计算评估seed序列覆盖的指标,并据此对seed序列进行次级筛选;
19.d)将read序列与次级筛选得到的seed序列比对,seed序列比对过程中相互间竞争reads,计算比对中seed序列的read覆盖度指标,逐步迭代去除read覆盖度指标不满足要求的seed序列,得到叁级筛选seed序列;
20.e)合并所述叁级筛选seed序列,并将reads与之进行比对,逐步迭代筛选得到满足要求的参考序列,其中逐步迭代筛选所用阈值相对于步骤d)更加严格;
21.f)根据步骤e)得到的所述参考序列及其reads数量,计算物种水平的reads含量及其占比;计算时将同属于一个物种的多个参考序列的reads数目加和得到该物种的reads
数,然后根据每个物种reads数目除以样本所含物种的reads数量的总和计算样本中每个物种的占比即丰度。
22.本发明还涉及一种通过测序获取微生物物种及相关信息的装置,所述装置包括:
23.测序数据获取模块,其用于获取测序数据,所述测序数据由靶向富集微生物特征核酸序列后经下一代测序技术对扩增产物进行测序得到;
24.特征序列数据库构建模块,其用于对所述测序数据进行聚类处理得到特征序列数据库,所述特征序列数据库包含一层或多层级的cluster,每个层级cluster中有至少一个子seed,最底层的cluster中有一或若干作为参考序列的seed;
25.比对分析模块,其用于将所述测序数据与所述特征序列数据库进行比对分析以鉴定所述被检测样本中的微生物组成,所述比对分析为如上所述方法中的所述比对分析所定义。
26.根据本发明的再一方面,本发明还涉及一种计算机可读存储介质,所述计算机存储介质用于存储计算机指令、程序、代码集或指令集,当其在计算机上运行时,使得计算机执行如上所述的方法中的步骤ii)。
27.根据本发明的再一方面,本发明还涉及一种电子设备,包括:
28.一个或多个处理器;以及
29.存储装置,存储一个或多个程序,
30.当所述一个或多个程序被所述一个或多个处理器执行,使得所述一个或多个处理器实现如上所述的方法中的步骤ii)。
31.本发明还涉及如上所述的方法、或如上所述的装置、或如上所述的计算机可读存储介质、或如上所述的电子设备在鉴定微生物中的应用。
32.1.尽管经过长期努力,现有技术仍然无法满意地解决应用短读长ngs技术完成基于诸如16s rrna基因序列等进化上高度保守的长片段序列信息进行微生物物种识别的问题。本发明很好地解决了该问题。对实验室和临床样本的检测证实,本发明实现了运用短读长ngs技术完成对诸如16s rrna基因序列等高度相似的长片段序列的准确鉴别,克服了以往靶向测序仅能够用于检测短序列的困难,实现了基于短片段测序在物种水平或更高分辨率下的微生物鉴定。
33.2.本发明可以正确地鉴定样本中存在的微生物物种、并测量各物种间数量上的相对比例,具有比现有技术更高的准确性与灵敏度。以细菌为例:对于单个物种的检测下限可以低至10cfu。本发明能够同时正确地检出多个(如5个或更多)物种混合样本中的全部微生物。即使两个物种间含量相差16倍或更多,本发明也能够同时正确地全部检出。
34.3.在对临床样本的检测中,实施例三至九中所有样本的平均测序数据量为55,663条reads,远低于目前mngs方法检测所需的数据量(10,000,000-100,000,000条reads)。全部样本中可用于微生物鉴定的有效数据量均在90%以上,而现有报道中mngs方法的有效数据通常占比为0.00001-0.01%。相比mngs方法,本发明展现了极高的数据效率。因而,本发明的检测成本远低于当前技术mngs的检测成本。
35.4.较高的测序覆盖深度才能保证检测的准确性。本发明检测中对靶序列覆盖率近100%、测序深度达到20
×
以上的微生物物种鉴定结果才能予以确认。但目前的公开报道中,mngs对受检微生物基因组的覆盖率要求可低至10%以内,平均测序深度自然低于1
×
。
因此,本发明应用于微生物检测的灵敏度和特异性均高于现有技术。
36.5.通过对人工模拟样本与临床样本的检测证实,本发明在保证低成本的同时,检测的准确性和灵敏度亦满足实际需要。因而,在保持mngs方法检测靶标广泛、受影响因素较少等优势的同时,本发明具备更高的检测灵敏度与准确性。
附图说明
37.为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
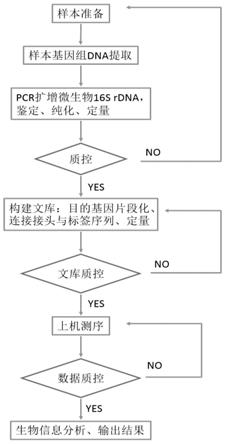
38.图1为本发明一个实施例中以基于原核生物16s rrna基因测序的微生物检测为例的检测流程示意图;
39.图2为本发明一个实施例中以基于16s rdna参考序列数据库的构建的主要操作流程的示意图;
40.图3为本发明一个实施例中构建了包含两层聚类层级结构的数据库的示意图;
41.图4为本发明一个实施例中参考序列数据库的seed聚类层级关系的示意图;
42.图5为本发明一个实施例中利用测序数据进行微生物物种鉴定的算法流程示意图。
具体实施方式
43.现将详细地提供本发明实施方式的参考,其一个或多个实例描述于下文。提供每一实例作为解释而非限制本发明。实际上,对本领域技术人员而言,显而易见的是,可以对本发明进行多种修改和变化而不背离本发明的范围或精神。例如,作为一个实施方式的部分而说明或描述的特征可以用于另一实施方式中,来产生更进一步的实施方式。
44.因此,旨在本发明覆盖落入所附权利要求的范围及其等同范围中的此类修改和变化。本发明的其它对象、特征和方面公开于以下详细描述中或从中是显而易见的。本领域普通技术人员应理解本讨论仅是示例性实施方式的描述,而非意在限制本发明更广阔的方面。
45.本发明涉及一种通过测序获取微生物物种及相关信息的方法,包括:
46.i)获取测序数据,所述测序数据由靶向富集微生物特征核酸序列后经下一代测序技术对扩增产物进行测序得到;
47.ii)将所述测序数据与特征序列数据库进行比对分析以鉴定所述待检测样本中的微生物组成;
48.其中所述特征序列数据库预先根据含有所述特征序列的参考序列的相似性做聚类处理,得到一层或多层级的cluster,每个层级cluster中有至少一个子seed,最底层的cluster中有一或若干作为参考序列的seed。
49.所述微生物包括细菌、古菌、真菌、支原体、衣原体、力克次氏体、螺旋体、和病毒,其中rna病毒的特征核酸序列可通过逆转录其rna基因组生成cdna后获得。
50.技术术语
51.本发明中涉及的技术术语包括如下:
52.ngs:next generation sequencing,下一代测序/第二代测序,又名高通量测序;
53.mngs:metagenomics next generation sequencing,宏基因组下一代测序;
54.its:internal transcribed spacer,内转录间隔区,是位于真菌核糖体rna(rrna)基因转录区或对应多顺反子rrna前体中大、小亚基rrna之间的核酸序列。
55.reads:测序读段,即高通量测序时产生的检测对象的一段序列;
56.cor:pearson’s correlation coefficient,皮尔逊相关系数;
57.nrmse:normalized root mean square error,标准化的均方根误差;
58.cv:coefficient of variation,变异系数;
59.alignment:序列比对;
60.reads mismatch rate:序列比对错误率;
61.fastq:存储序列及其质量值的四行的文本文件格式;
62.adapter:测序中使用的接头序列;
63.cluster:类、簇;
64.seed:种子,也即类中心;依据所在cluster的层级不同,seed又分为子seed与作为参考序列的seed,但二者可为“seed”所概括;
65.bowtie2:一个将短序列比对到大基因组的比对软件;
66.mean depth:平均测序深度;
67.gap:空白、断点,此处代表无覆盖;
68.end gap:末端的无覆盖;
69.middle gap:中间段无覆盖;
70.em:expectation maximization,期望最大化;
71.overlap graphs:用于表征多个核酸序列间在序列编码上相互重叠关系的图;
72.paired-end reads:双端序列,从目前片段的正反向分别测序产生的序列;
73.de novo组装:从头组装,将小片段组装成较长片段的方法;
74.heuristic:经验性的。
75.参考序列:在本发明中,若无特殊说明,参考序列是指能够表明微生物种属的特征序列,其通常是保守的,参考序列一般包括16s rrna基因、18s rrna基因、its核酸序列、rna病毒的rna为模板的rna聚合酶基因(rdrp)、病毒衣壳蛋白编码基因、逆转录病毒的pol基因等或者其他可以反映微生物种属特征的核酸序列中的一种或多种的全长序列。
76.检测的样本及其预处理,测序文库的建立
77.在本发明中,检测的对象可以来自于生物(微生物宿主)或来自于含有微生物的环境样本。在一些实施方式中,所述待检测样本为来自于微生物宿主的样本或来自于含有微生物的环境样本:所述来自于微生物宿主的样本优选包括但是不限于:粪便、肠道内容物、皮肤、组织、痰液、血液、唾液、牙菌斑、尿液、阴道分泌物、胆汁、支气管肺泡灌洗液、脑脊液、胸水、腹水、盆腔积液、脓液以及瘤胃中的至少一种;在一些实施方式中,所述来自于含有微生物的环境样本优选包括:物体内外表面、生活用水、医疗用水、工业用水、食品、饮料、肥料、废水、火山灰、冻土层、淤泥、土壤、堆肥、污染河流养殖水体以及空气中的至少一种。
78.在一些实施方式中,所述宿主是动物;进一步可以选择包括人类及所有畜养(如家
畜和宠物)和野生的动物及禽鸟,其非限制性地包括牛、马、乳牛、猪、绵羊、山羊、大鼠、小鼠、狗、猫、兔、骆驼、驴、鹿、貂、鸡、鸭、鹅、火鸡、斗鸡等。
79.以基于原核生物16s rrna基因测序的微生物检测为例,样本的检测流程示意图如图1所示,详述如下:
80.样本的预处理:针对不同类型、来源的样本,可能需要对样本进行预处理,以适应核酸提取的需要。处理方式包括但是不限于使用无菌水、ddh2o(双蒸馏水)、无菌生理盐水、无菌pbs(磷酸缓冲盐溶液)等液体对样本进行洗涤,使用过滤、离心等方式对样本进行浓缩,使用梯度离心等方式对样本中的某些成分进行分离,或者使用某些满足实验需求的试剂盒对样本中的某些成分进行分离、或者对样本中某一部分的核酸进行去除或者富集。
81.核酸的提取:使用核酸提取试剂盒,提取经过预处理后样本中包含的全部核酸物质。使用的核酸提取试剂盒不局限于某一个厂家,也不局限于某一种方法,能获得满足实验需要的质量要求的核酸物质即可。所提取的核酸包括dna、rna,或者两者同时提取。在此步骤开始之前,可以向样本中加入一定量的已知序列的核酸,该核酸序列满足以下条件:1)可以在下一步骤配制的反应体系下被扩增;2)可以被下一步骤加入的已有引物或者单独准备的引物相匹配;3)全部序列已知;4)所使用的序列不会干扰对样本中可能存在的任何目的序列的分析;5)核酸序列可以单独存在,也可以依赖于质粒、病毒、细胞等载体存在;6)所加入的核酸序列可以被此步骤的操作所获得,并且存在于最终提取到的核酸物质中。在本技术方案中,加入已知序列的核酸可以帮助更好的判断检测结果中由于采样、实验等环节引入的污染,但是并不是本技术方案的必须。不加入已知序列的核酸不会影响本技术方案的完整性,加入已知序列的核酸也不构成对本技术方案的创新。加入已知序列的核酸也不局限于在此步骤进行。
82.特定核酸序列的靶向富集:使用某些方法对能够提供微生物分类信息的核酸序列进行富集,使这些序列的核酸在样本的全部核酸序列中占据更高的比例,并且对富集产物进行纯化和定量。富集的方法包括但是不限于pcr、杂交捕获等方式。对富集产物的纯化包括但是不限于吸附柱纯化、磁珠纯化等方式,目的是去除富集处理过程在样本中残留的酶、引物、探针、盐、金属离子等成分,获得纯净的较长片段(大于20bp)的核酸序列。定量是为了确定得到样本中核酸的浓度,进而根据体积计算样本中的核酸含量。定量方式包括紫外分光光度法、染料结合法等方法。富集的目标序列,是目前常用的对微生物进行物种分类的序列,对于原核生物,可能是核糖体16s rrna基因序列;对于真核生物,可能是核糖体18s rrna基因序列或者its序列;对于病毒,可能是其基因组中同时具备进化保守性和物种特异性的核酸序列,如冠状病毒基因组中的pol(rna-dependent rna polymerase)和n(nucleocapsid)基因。这类序列通常较长,并且可能具有不同物种之间高度相似的部分,使用现有的短读长ngs测序和数据分析方法不能正确区分物种来源。对于获取的rna,还可能需要先进行反转录,将rna反转录为cdna,然后再进行特定序列的靶向富集。在此步骤,配制反应体系时还可以加入一定量的已知序列的核酸(作为阳性对照),该核酸序列满足以下条件:
83.1)可以在本步骤配制的反应体系下被扩增;
84.2)可以被本步骤加入的已有引物或者单独准备的引物相匹配;
85.3)全部序列已知;
86.4)所使用的序列不会干扰对样本中可能存在的任何目的序列的分析;
87.5)核酸序列可以单独存在,也可以依赖于质粒、病毒、细胞等载体存在。
88.在一些实施方式中,在核酸富集之后还包括一步或多步质检流程,其目的是检测对目标核酸序列富集的效果。质量控制的手段包括对核酸含量的检测、核酸纯度的检测、以及富集后的核酸序列的片段长度检测等。经过有效富集的样本,会有更高的测序检测效率,通常也意味着样本中微生物的含量更加丰富。通过对富集效率的判断,可以预测样本中微生物的含量水平。对于富集质量不符合预期的样本,可以重新进行富集操作,但并不都是必须重新富集,富集质量不符合预期的样本也可以继续进行后续实验。
89.测序文库构建:目的是将富集得到的核酸,转换为ngs平台可以检测的核酸短片段。主要步骤是将长的核酸序列打断为ngs平台可以读取的长度,同时在片段两端加入相应的测序引物,使得测序仪可以对核酸序列进行检测。如果加入的测序引物含有标签(barcode/index),还可以对样本来源进行区分。在此步骤开始之前,在上一步获得的核酸中可以加入一定量的已知序列的核酸,该核酸序列满足以下条件:
90.1)全部序列已知;
91.2)所使用的序列不会干扰对样本中可能存在的任何目的序列的分析;
92.3)核酸序列可以单独存在,也可以依赖于质粒、病毒、细胞等载体存在。
93.在本发明中,加入已知序列的核酸可以帮助更好的判断检测结果中由于采样、实验等环节引入的污染,但是并不是本发明的必须。不加入已知序列的核酸不会影响本技术方案的完整性,加入已知序列的核酸也不构成对本技术方案的创新。加入已知序列的核酸也不局限于在此步骤进行。
94.测序文库构建具体实验包括以下步骤:
95.1)将长的核酸序列片段打断为较短的核酸序列片段。由于提取到或者富集之后的核酸序列仍然很长,大大超过ngs仪器的读取能力,需要将长片段打断为ngs仪器可以读取的长度的短片段,然后同时进行检测,才能够获得全部的核酸序列信息。打断的方法包括但是不限于物理方法(例如使用超声设备使得核酸断裂)、生物学方法(例如使用核酸酶、转座酶等方法将核酸序列切断)等。
96.2)进行末端修复。在文库构建的整个环节中,作为操作对象的核酸都是双链dna(dsdna)。将长片段dsdna打断之后生成的短片段dsdna的两个末端不会很整齐,通常一条链会有几个碱基的突出,形成粘性末端。根据不同测序平台的设计,需要对打断后的dna进行不同方式的修复。例如,如果使用thermofisher的ion torrent测序平台,需要将末端修复为完全齐平的形式,如果使用illumina测序平台,需要将末端修复为其中一条链有一个额外的腺嘌呤(a)的形式。
97.3)片段筛选。使用片段筛选磁珠,对样本中的核酸片段进行筛选,仅保留适宜长度的核酸片段,过长或者过短的核酸片段被丢弃。核酸片段的长度根据所选用的测序平台、测序试剂、测序条件的不同而不同。
98.4)连接测序接头。测序接头是两段具有特定序列的dsdna。在测序仪器中,测序反应需要从这些特定序列开始工作,测序引物可以含有或者不含barcode/index序列,barcode/index序列可以用于区分同一次测序实验中不同样本来源的序列。使用t4连接酶等酶类工具,将两个测序引物分别连接在经过末端修复的短片段dsdna的两端。只有两端分
别连接了一端一个测序引物的dsdna才能够被测序。
99.5)文库富集。一个样本中所有的正确连接了测序引物的待测核酸序列称为测序文库。文库富集是使用一定方法,通常是pcr,将正确连接了测序引物的核酸序列数量放大,增加其拷贝数,方便后续工作。富集步骤在实验流程中并不总是必须的。
100.6)在本环节的每一步反应之后,都有相应的纯化操作,目的是将核酸与反应试剂分离,获得纯净的核酸进入下一步反应。随着试剂与反应条件的调整,并不是每一次的纯化操作都是必须的,这样的调整不超出本技术方案的范围,纯化所用的试剂与方法也不构成对本技术方案的限制。
101.在一些实施方式中,测序文库构建后还包括质控步骤,其目的是检测构建的测序文库是否符合测序要求。质量控制的手段包括对核酸含量的检测、核酸纯度的检测、以及富集后的核酸序列的片段长度检测等。只有片段长度符合测序仪器要求、含量足够、纯度合格的文库才能用于后续测序。本次质控是实验流程中保障实验质量的一个环节,控制参数与所选用的测序平台有关,但是并不是本技术方案所要求的必须环节。
102.上机测序:根据所选用的测序仪厂家、型号、试剂的说明书进行实验。本技术所兼容的ngs测序仪厂家包括但不限于thermo fisher,illumina,bgi等主流厂家所有目前在市场销售的仪器与试剂。
103.数据分析流程
104.本发明中数据分析流程的逻辑是,将ngs测序获得的短序列片段(reads)比对(map)到微生物基因组特征序列数据库中的所有参考序列上;计算每个参考序列上reads实际覆盖的统计特征参数如cv等,并将每个参考序列上reads实际覆盖与(如果该参考序列代表的微生物包含在样本中时)理论预测的reads覆盖相比较,该理论预测的reads覆盖可根据数学模型如概率和统计模型、或经验/实验数据构造。比较的结果体现为多个不同的比较表征参数(如nrmse等);根据统计特征参数和比较表征参数是否符合要求的标准,将达不到标准的参考序列淘汰。循环重复该筛选操作,直至所有剩余的参考序列的统计特征参数和表征参数均满足既定的最终标准,则这些参考序列所代表的微生物物种即为物种检测的结果。计算这些参考序列各自比对上的reads数量在该样本比对上参考序列的总reads数量中所占的比例,即为各参考序列所代表的微生物物种在样本微生物总数量中的占比。
105.上述基于每个参考序列上reads实际覆盖的统计特征参数和比较表征参数的参考序列筛选方法仅是其中可以使用的筛选方法之一,其他适合的筛选方法也可采用,例如,仅使用统计特征参数或比较表征参数进行筛选;或使用基于fisher’s exact test富集分析的方法进行筛选;或基于贝叶斯概率(bayesian probability)的em算法,等等。
106.本发明根据数据库所含参考序列之间的序列相似性,对所有参考序列进行聚类,并为每个类(cluster)挑选出具代表性的参考序列即seed;对所有的seed进行聚类操作,并为生成的每个cluster选出代表性的参考序列即seed id;可根据需要在seed id的基础上做进一步的聚类操作和选取seed id。该操作可根据需要进行多次。最终构建出一个多层级聚类树(hierarchical clustering tree)结构(该树结构可以有或没有根(root)),其中树的叶子(leaf)由微生物基因组特征序列数据库的参考序列组成、构成树结构的最底层,叶子上方的第一层节点(node)为第一层cluster的seed,其余各层的节点分别为代表其下方连接的所有节点构成的cluster的seed id。对数据库参考序列的筛选可以从聚类树的最上
层节点开始,逐层向下进行。
107.在一些实施方式中,所述比对分析包括:
108.a)去除所述测序数据中的低质量和含有测序接头序列的reads;
109.b)将reads序列与所述特征序列数据库的seeds序列进行比对,将因pcr扩增而产生的完全重复reads去除,并对事件“有reads序列比对上”与seeds序列做统计学独立性检验,选出与事件“有reads序列比对上”相关的seeds序列,得到初级筛选seed序列;
110.c)将read序列与所述初级筛选seed序列进行比对,其中所述初级筛选seed序列之间不竞争reads,计算每个seed序列的reads覆盖情况,计算评估seed序列覆盖的指标,并据此对seed序列进行次级筛选;
111.d)将read序列与次级筛选得到的seed序列比对,seed序列比对过程中相互间竞争reads,计算比对中seed序列的read覆盖度指标,逐步迭代去除read覆盖度指标不满足要求的seed序列,得到叁级筛选seed序列;
112.e)合并所述叁级筛选seed序列,并将reads与之进行比对,逐步迭代筛选得到满足要求的参考序列,其中逐步迭代筛选所用阈值相对于步骤d)更加严格;
113.f)根据步骤e)得到的所述参考序列及其reads数量,计算物种水平的reads含量及其占比;计算时将同属于一个物种的多个参考序列的reads数目加和得到该物种的reads数,然后根据每个物种reads数目除以样本所含物种的reads数量的总和计算样本中每个物种的占比即丰度。
114.在一些实施方式中,所述待检测样本为来自于微生物宿主的样本,步骤a)还包括:去除所述样本中所述宿主的核酸测序数据干扰。
115.在一些实施方式中,在步骤d)中,所述逐步迭代去除read覆盖度指标不满足要求的seed序列之后还包括cluster内部参考序列的筛选步骤:
116.将上步获得的每一个seed所属cluster的参考序列与reads进行比对,相同cluster内部的参考序列间竞争reads;统计每个参考序列的reads覆盖情况,根据reads覆盖指标进行过滤,逐轮迭代去除reads覆盖不佳的seed序列,其中逐轮迭代筛选所用阈值相对于步骤d)前面所述步骤更加严格。
117.在一些实施方式中,在步骤f)后还包括步骤g):排除实验环境中的背景污染物种的核酸测序数据干扰。
118.在一些实施方式中,在步骤b)中,所述统计学独立性检验为费舍尔精确检验,其具体包括:
119.将每个比对上的reads数量大于一定数量的参考序列记为“有read比对”,否则记为“无read比对”;根据参考序列数据库中seeds的聚类层级关系,统计检验聚类树中每个seed下属的叶子节点中是否显著富集“有read比对”的seed id,逐级筛选出满足要求的seeds。
120.在一些实施方式中,所述特征序列数据库的构建方法包括:
121.获取含有所述特征序列的参考序列的公共数据库,去除所述数据库中所述参考序列在所述扩增引物之外的两端序列,得到第一数据库;
122.根据物种内相似性对所述第一数据库中存在模糊碱基的所述参考序列进行矫正处理,并根据物种注释以及相似性去除100%相似的冗余参考序列,得到第二数据库;
123.根据所述第二数据库中所述参考序列的相似性对其做聚类处理。
124.在一些实施方式中,在构建所述第一数据库时,去除所述数据库中所述参考序列在所述扩增引物之外的两端序列和引物序列。
125.在一些实施方式中,在构建所述第二数据库时,还包括:
126.将每条参考序列与ncbi nt/nr库进行blast比对,按照序列相似性和/或覆盖度的规则从ncbi nt/nr库中筛选出匹配的参考序列集;
127.在所述匹配的参考序列集的物种注释中选择最具代表性的物种分类,并使用该分类信息校正所针对的参考序列的物种注释。
128.在一些实施方式中,所述聚类处理包括第一聚类处理:
129.将所有非冗余的参考序列按照相似性进行聚类;
130.或;
131.1)按照物种内非冗余的参考序列的相似性进行聚类;
132.2)对1)中得到的seed按照相似性进行聚类,将聚在同一个类里的不同物种的seed及其子序列合并,然后按照99.5%相似性进行重聚类,用新获得的cluster替换参与重聚类计算的旧cluster。
133.在一些实施方式中,所述聚类处理还包括第二聚类处理:
134.在cluster中seed的子序列太多的情况下,按照比所述第一聚类处理更高的相似性标准对所述第一聚类处理得到cluster进行拆分,用拆分后形成的新cluster替换掉拆分前的cluster。
135.在一些实施方式中,所述聚类处理还包括第三聚类处理:
136.按照不同的相似度阈值,对所述第二聚类处理得到的cluster的seed参考序列进行层级聚类,创建一个有层次的嵌套的树。
137.下面以基于原核生物16s rrna基因ngs测序的微生物检测为例,对数据分析流程加以详细说明。
138.数据分析流程主要包括两大部分的内容,1)分析流程中使用的16s rrna基因(即rdna)参考序列数据库构建方法;2)利用测序数据进行数据分析的算法流程。
139.1.16s rdna参考序列数据库的构建,主要操作流程的示意图如图2所示。
140.1)下载公共数据库中最新的16s rdna参考序列数据,保证数据库序列来源的完备性。下载数据库包括但不限于ncbi及silva数据库,数据库包含的参考序列种类和数量可根据具体需要选择,例如,可包含100、250、500、1000、2000、10,000种、甚至全部已有种类等等不同的数量。
141.2)去除参考序列中的富集扩增引物以及两端引物之外的序列
142.本步骤的目的是获取不包含引物的参考序列。可以使用基于smith-waterman局部比对算法或者其他比对定位短序列的方法,根据一定的相似性(例如但不限于80%、85%、90%、95%等)匹配富集时使用的扩增引物(本步骤中简称扩增引物)信息及在参考序列中定位富集扩增引物,然后根据引物匹配的位置进行序列剪切,去除扩增引物以及引物之外的两端序列。另外,也可以仅仅去除引物以外的两端序列。
143.3)对16s rdna序列进行物种注释校正
144.由于数据库中部分参考序列的物种注释信息可能存在误差,为了确保物种注释信
息能够最大程度上反映其正确的物种归属,本技术方案对物种注释进行校正。但如果获取的参考序列物种注释信息足够确信,亦可不做此校正。校正流程:将每条参考序列与ncbi nt/nr库进行blast比对,按照序列相似性99.5%、覆盖度(coverage)99%(适当提高或者降低相似性值也在此技术范围内)的规则从ncbi nt/nr库中筛选出匹配的参考序列集。在匹配的参考序列集的物种注释中选择最具代表性的物种分类,并使用该分类信息校正所针对的参考序列的物种注释。同时根据该代表性物种分类在匹配的参考序列集的物种注释中的唯一性程度对所针对的参考序列的物种注释的确定性进行分级,物种注释的确定性级别包括:certain,limitedcertainty,rare annotation(按确定性由高到低排序),也可以根据实际确定性情况进行更多/少级别的的分类,甚至是不分类。
145.4)对16s rdna序列中的模糊碱基进行校正
146.数据库中某些参考序列的部分位置的碱基不明确或包含简并碱基。为了明确这些位置最具代表性的碱基,我们按照规则进行校正,主要步骤是:对在同一个物种内相似性大于97%的参考序列使用muscle进行多重序列比对,对于模糊碱基位置使用最具代表性的碱基进行替换。此步骤之后若还存在模糊碱基,则选择其同属中具有97%相似性的参考序列进行多重序列比对,对于模糊碱基位置使用最具代表性的碱基进行替换。处理之后依然存在的模糊碱基予以保留。
147.5)根据物种注释以及相似性对数据库进行去冗余
148.本步骤主要是为了去除100%相似的冗余参考序列,同时确保需要的参考序列不被误去除。因此,本技术方案要求被去除的参考序列需被其他参考序列所包含、以及物种注释的确定性等级较低,同时,还结合序列两端的引物匹配情况进行判断,比如:一个参考序列两端引物完整,但是却被另外一个参考序列完全包含,这种情况下前一个参考序列需保留;又或者,如果较短的参考序列的物种确定性等级更高,那么短的参考序列也可以保留。还有一些更复杂的情况比如参考序列某包含引物的端被另一个更长的参考序列包含的处理等。
149.6)对物种内非冗余的参考序列按照指定阈值的相似性进行聚类
150.聚类的目的是,一方面分散后续分析的计算压力,另一方面减少比对时序列之间的竞争,从而能更准确的逐步挑选出合适的参考序列。按照相似性99.5%(相似性指标可根据实际数据库参考序列的情况进行适当调整)的标准,对每个物种内所有的非冗余参考序列进行聚类。聚类后,每个参考序列类(cluster)中的代表序列作为该类的seed,与它聚在同一个cluster里的其他序列作为其子序列。
151.7)对物种间相似性较高的类进行合并后按照指定阈值的相似性重聚类
152.对6)中得到的所有seed按照99%(可根据6)中seed聚类的相似性指标作出适当调整)相似性进行聚类,将聚在同一个类里的不同物种的seed及其子序列合并,然后按照99.5%相似性进行重聚类,用新获得的cluster替换参与重聚类计算的旧cluster。另外,本步骤可以和6)合并,直接使用所有的序列进行聚类,而非按照前面描述的先物种内聚类、再合并物种间相似的类。但如果距离较近的物种太多,则直接做聚类的方式并不一定能达到很好的分析效果。
153.8)将聚类后较大的cluster按照更高相似性进行拆分
154.在cluster中seed的子序列太多的情况下,按照更高相似性(例如99.6%、99.8%、
99.9%等)标准对该cluster进行拆分,用拆分后形成的新cluster替换掉拆分前的cluster。至此,构建了包含两层聚类层级结构的数据库:第一层由cluster的seed参考序列组成,第二层包含所有cluster的子参考序列。其结构示意图如图3所示。
155.9)对构建的cluster的seed参考序列进行层级聚类
156.依次按照97%、98%、99%(仅以此3种聚类相似性指标为代表,只要能达到逐层聚类的效果都可以)的相似性对所有seed参考序列进行聚类。主要步骤为:首先对所有的seed参考序列按照97%相似性聚类,得到第一层的cluster(包含seed及其子序列);然后针对每个cluster分别对其内部的参考序列按照98%相似性进行聚类,得到第二层的cluster;之后对第二层的每个cluster分别对其内部的参考序列按照99%相似性进行聚类,得到第三层的cluster。最终构建出包含数据库中所有seed参考序列的索引层级结构,使得每个seed参考序列获得分别对应于三种不同序列相似性聚类的seed id。至此,所有参考序列的索引信息构成了一个由四列信息组成的文件。参考序列数据库的seed聚类层级关系的示意图如图4所示。
157.2.利用测序数据进行微生物物种鉴定的算法流程,具体流程如图5所示。
158.1)测序数据的质量控制
159.对进入分析流程的fastq格式的测序短序列(read)数据进行筛选,过滤掉低质量和含有adapter序列的reads。
160.2)去除宿主(人)的核酸序列污染
161.使用bowtie2(或其他短序列比对工具)按默认参数将所有read序列与人类参考基因组hg19或hg38序列比对,过滤掉比对上人基因组序列的read序列;
162.3)数据库seed序列的初级筛选
163.使用hisat2(比对工具不限,能高效地实现reads与参考序列较准确匹配的目的即可)将read序列与数据库的seed序列进行比对。根据层级聚类信息,利用富集分析原理对seed进行初级筛选。具体流程如下:
164.使用hisat2软件,将所有reads序列与数据库所有seed序列进行比对。以低于某个阈值(如0.5%、1%、1.5%,2%等)的read mismatch rate为条件过滤比对结果,并将alignment中因pcr重复而产生的reads去除。将每个比对上的reads数量大于一定数量(例如5、10、15等条)的参考序列记为“有read比对”,否则记为“无read比对”。根据参考序列数据库中seeds的聚类层级关系即聚类树(见图4),利用fisher’s exacttest进行富集分析,统计检验聚类树中每个seed下属的叶子节点中是否显著富集“有read比对”的seed id,逐级筛选出满足要求的seeds。
165.fisher’s exact test富集分析说明如下:
166.根据每个seed的情况,为其构建下表,并按表下所列公式计算fisher’s exact test检验的概率值。表格中为同时满足行和列条件的seed id计数。当p值小于或等于某个阈值(如0.001,0.01,0.05,0.1,0.2,0.3,0.4,0.5等)时,该被检验的seed下属的聚类中显著地富集“有read比对”,且p值越小富集越显著。
[0167][0168][0169]
4)数据库seed序列的次级筛选
[0170]
使用bowtie2(比对工具不限,能达到reads与参考序列较准确匹配的目的即可,例如其他序列比对工具maq、soap、bwa、novoalign等)将read序列与初级筛选出的seed序列进行比对(seed序列间不竞争reads),计算每个seed序列的reads覆盖情况,计算评估seed序列覆盖的指标,并据此对seed序列进行次级筛选。具体计算流程如下:
[0171]
使用bowtie2将read序列与初级筛选出的seed序列进行比对(主要是能得到比对相似度满足一定要求的alignment,ref间不竞争reads),按照阈值(如0.5%、1%、1.5%,2%等)的reads mismatch rate对alignment进行过滤,并将alignment中因pcr重复而产生的reads去除。计算每个seed序列的reads覆盖情况,计算指标包括cv(coefficient of variation),coverage(覆盖度),mean depth(平均测序深度),end gap(序列两端的覆盖缺口),middle gap(序列中间的覆盖缺口)等。根据相对宽松的指标对seed序列进行筛选,满足指标的seed序列通过次级筛选。
[0172]
5)数据库seed序列的叁级筛选
[0173]
将通过次级筛选的seed序列进行随机分组。在各组内使用bowtie2或其他序列比对工具例如maq、soap、bwa、novoalign等,将read序列与seed序列比对,seed序列比对过程中相互间竞争reads。计算alignment中seed序列的read覆盖度指标,逐步迭代去除read覆盖度指标不满足要求的seed序列。具体计算流程如下:
[0174]
将过滤后的seed进行随机分组,每组内使用bowtie2将read序列与seed序列比对。比对过程中,seed序列之间竞争reads,reads在比对得分排名靠前的seed序列间随机分配。然后按照某个阈值的reads mismatch rate对alignment进行过滤,并将alignment中因pcr重复而产生的reads去除。计算alignment的覆盖度指标,包括cor(pearson’s correlation coefficient,计算预期的seed序列的reads覆盖模型与alignment获得的实际覆盖之间的一致度)、nrmse(normalized root mean square error,预期模型与实际覆盖间的差异度)、coverage、mean depth、end gap、middle gap等。依据alignment的覆盖度指标是否满足设定的参数,例如mean depth≥15或20、end gap≤30或40、nrmse≤0.4或0.35、middle gap≤0或5或10或15,等等,其中mean depth计算的时候从整个物种水平进行计算的,即属于同一个物种的多个seed深度之和满足参数即可。参数越严格进入后续分析的seed可能会越少,如果参数较严格的情况下覆盖尚且良好的seed能作为最终候选参考序列的可能性也较大。将不满足要求的seed序列过滤掉,直到剩余的seed序列全部满足要求为止。将各组筛选得到的seed序列合并,进入下一步流程。
[0175]
6)cluster内部参考序列的筛选
[0176]
前几步流程在数据库的seed层进行筛选。为了得到更准确的符合要求的参考序
列,本步骤对叁级筛选得到的每个seed所属的cluster的子序列进行筛选。另外,此步筛选也可调整为:如果5)筛选出的seed序列的覆盖足够好,可直接进入最终的步骤候选。仅对reads覆盖不够好的seed进行cluster内部比对筛选。下面以前一种方法为例进行详细说明。
[0177]
使用bowtie2(比对工具不限,能达到reads与参考序列较准确匹配的目的即可)将reads序列与叁级筛选获得的每一个seed所属cluster的参考序列(包含seed序列及其子序列)进行比对。参考序列间竞争reads。统计每个参考序列的reads覆盖情况,根据reads覆盖指标进行过滤,逐轮迭代去除reads覆盖不佳的参考序列。具体计算流程如下:
[0178]
分别对筛选出的每一个seed序列所属的cluster的参考序列使用bowtie2与reads序列进行比对。采用较前面几步更严格的比对参数,并允许参考序列间竞争reads。初始迭代时,如reads比对上多个参考序列且得分都是排名最高(top1),则reads在这些参考序列间随机分配。后续迭代过程中,比对得分top1的参考序列共享的reads的分配将根据各参考序列在前一轮比对之后得到的read count和覆盖度指标值按比例进行。read count越多和覆盖度指标越好,该参考序列获得reads的概率就越大。比对完成后,按照相比前几步更严格阈值的reads mismatch rate对alignment进行过滤。对alignment中有mismatch/indel的位点,根据优势碱基占比情况去除可能是错误匹配的reads。将alignment中因pcr重复而产生的reads去除。计算reads覆盖度指标,包括cor(计算预期的reads覆盖模型与实际reads覆盖间的一致度)、nrmse(计算预期的reads覆盖模型与实际reads覆盖间的差异度)、coverage、mean depth、end gap、middle gap等。依据reads覆盖度指标是否满足设定的参数,将不满足要求的参考序列过滤掉。每一轮迭代最多过滤掉参考序列总数的1%、或5%、或10%、或15%、或30%等(比例可按照所需的收敛速度进行调整),直到剩余的参考序列全部满足要求。而后对同属一个物种的多个参考序列进行去重复计算。使用muscle(或其他序列比对工具例如clustalw、t-coffee、mafft等)对相关参考序列进行两两比对,将比对位置完全一致的两个参考序列中reads覆盖较差的一方去除。最后将每个seed所属cluster内部筛选得到的参考序列作为候选参考序列进入下一步流程。
[0179]
7)参考序列的终极筛选
[0180]
合并cluster内部筛选获得的参考序列,并使用bowtie2(或其他序列比对工具例如maq、soap、bwa、novoalign等进行合理参数设置)将reads与参考序列进行比对。使用更加严格的参数,逐步迭代筛选得到最终满足要求的参考序列。具体计算流程如下:
[0181]
合并cluster内部筛选获得的参考序列,使用bowtie2将reads与参考序列进行比对。比对、筛选、以及迭代过程均与6)相同,但覆盖筛选参数较6)更严格。完成6)的全部操作后,根据每个参考序列拥有的独有reads的数量情况,对参考序列进行最后的过滤。如果一个参考序列与另一参考序列间的两两指定区域内(末端序列不考虑)比对位置完全一致,则根据序列相似性和各自拥有的独有reads的数量情况,将独有reads数量较少(数量差距需要满足一定的范围,例如差异倍数超过1.5、2、2.5等)一方去除。
[0182]
8)计算样本所含微生物各物种间的数量比例
[0183]
根据7)得到的样本中所有的参考序列及其reads数量,计算物种水平的reads含量及其占比。将同属于一个物种的多个参考序列的reads数目加和得到该物种的reads数,然后计算样本中每个物种的占比即每个物种reads数目除以样本所含物种的reads数量的总
和。根据每个参考序列的各统计指标(包括meandepth、nrmse、cor、gap),使用logistic回归模型计算每个参考序列的可信度,其中模型的训练数据来源于多批次的实验分析结果。
[0184]
9)背景污染物种的排除
[0185]
由于采样或实验环境(包括实验使用的试剂和耗材)中存在各种类型不一的环境微生物,从而构成微生物检测结果中的背景污染。本技术方案去除背景污染的方法包括两个步骤:第一步,计算物种占比以及判断该物种是否在重要的临床致病物种列表中,即占比很低且不在重要临床致病物种列表中的物种将被当作假阳性而排除;第二步,过滤在阴性对照样本中出现的物种。但由于临床样本也可能确实包含环境中存在的某些微生物物种,所以不能简单地直接排除。为此,实验中设置了多个阴性对照样本,同时每个样本中(包括被测样本和对照样本)引入一种内部参照品(internal control),用于对同一样本中物种的定量进行归一化。去除背景污染物种的主要方法是:分别计算各临床样本和对照样本中该物种reads的归一化含量,然后计算该物种在临床样本中的含量是一个来自该物种(或者所有对照样本中检测到的物种)在阴性对照样本中含量的统计分布(如正态分布、poisson分布、weibull分布、或其他已知的理论分布、或基于数据的重抽样(如bootstrapping、jackknife等方法)获得的经验分布)的样本的概率。如概率大于某个阈值(如0.001,0.01,0.05,0.1,0.2,0.3,0.4,0.5等),则认为该物种是背景污染并从检测结果中去除,否则予以保留。
[0186]
至此,基于测序reads数据对参考序列数据库的筛选流程获得了最终满足要求的参考序列。根据所获参考序列的物种分类注释信息,可以获得样本所含微生物的物种构成。
[0187]
微生物测序数据分析装置、计算机可读存储介质及电子设备
[0188]
本发明还涉及一种微生物测序数据分析装置,所述装置包括:
[0189]
测序数据获取模块,其用于获取测序数据,所述测序数据由靶向富集微生物特征核酸序列后经下一代测序技术对扩增产物进行测序得到;
[0190]
特征序列数据库构建模块,其用于对所述测序数据进行聚类处理得到特征序列数据库,所述聚类处理为如上所述方法中的所述聚类处理所定义;
[0191]
比对分析模块,其用于将所述测序数据与所述特征序列数据库进行比对分析以鉴定所述待检测样本中的微生物组成,所述比对分析为如上所述方法中的所述比对分析所定义。
[0192]
在一个实施方式中,所述装置包括:
[0193]
测序数据获取模块,其用于获取测序数据,所述测序数据由引物对微生物特征序列进行靶向富集后经下一代测序技术对扩增产物进行测序得到;
[0194]
特征序列数据库构建模块,其用于对所述测序数据进行聚类处理得到特征序列数据库,所述特征序列数据库包含一层或多层级的cluster,每个层级cluster中有至少一个子seed,最底层的cluster中有若干作为参考序列的seed;
[0195]
比对分析模块,其用于将所述测序数据与所述特征序列数据库进行比对分析以鉴定所述待检测样本中的微生物组成,所述比对分析模块包括:
[0196]
a)第一模块,其用于去除所述测序数据中的低质量和含有测序接头序列的reads;
[0197]
b)第二模块,其用于将reads序列与所述特征序列数据库的seeds序列进行比对,将因pcr重复而产生的reads去除,并对reads序列与seeds序列做统计学独立性检验,选出
与reads序列相关的seeds序列,得到初级筛选seed序列;
[0198]
c)第三模块,其用于将read序列与所述初级筛选seed序列进行比对,其中所述初级筛选seed序列之间不竞争reads,计算每个seed序列的reads覆盖情况,计算评估seed序列覆盖的指标,并据此对seed序列进行次级筛选;
[0199]
d)第四模块,其用于将read序列与次级筛选得到的seed序列比对,seed序列比对过程中相互间竞争reads,计算比对中seed序列的read覆盖度指标,逐步迭代去除read覆盖度指标不满足要求的seed序列,得到叁级筛选seed序列;
[0200]
e)第五模块,其用于合并所述叁级筛选seed序列,并将reads与之进行比对,逐步迭代筛选得到满足要求的参考序列,其中逐步迭代筛选所用阈值相对于步骤d)更加严格;
[0201]
f)第六模块,其用于根据步骤e)得到的所述参考序列及其reads数量,计算物种水平的reads含量及其占比;计算时将同属于一个物种的多个参考序列的reads数目加和得到该物种的reads数,然后根据每个物种reads数目除以样本所含物种的reads数量的总和计算样本中每个物种的占比。
[0202]
本发明还涉及一种计算机可读存储介质,所述计算机存储介质用于存储计算机指令、程序、代码集或指令集,当其在计算机上运行时,使得计算机执行如上所述的方法中的步骤ii)。
[0203]
可以采用一个或多个计算机可读的介质的任意组合。计算机可读介质可以是计算机可读信号介质或者计算机可读存储介质。计算机可读存储介质例如可以是——但不限于——电、磁、光、电磁、红外线、或半导体的系统、装置或器件,或者任意以上的组合。计算机可读存储介质的更具体的例子(非穷举的列表)包括:具有一个或多个导线的电连接、便携式计算机磁盘、硬盘、随机存取存储器(ram)、只读存储器(rom)、可擦式可编程只读存储器(eprom或闪存)、光纤、便携式紧凑磁盘只读存储器、光存储器件、磁存储器件、或者上述的任意合适的组合。在本文件中,计算机可读存储介质可以是任何包含或存储程序的有形介质,该程序可以被指令执行系统、装置或者器件使用或者与其结合使用。
[0204]
计算机可读的信号介质可以包括在基带中或者作为载波一部分传播的数据信号,其中承载了计算机可读的程序代码。这种传播的数据信号可以采用多种形式,包括——但不限于——电磁信号、光信号或上述的任意合适的组合。计算机可读的信号介质还可以是计算机可读存储介质以外的任何计算机可读介质,该计算机可读介质可以发送、传播或者传输用于由指令执行系统、装置或者器件使用或者与其结合使用的程序。
[0205]
计算机可读介质上包含的程序代码可以用任何适当的介质传输,包括——但不限于——无线、电线、光缆、rf等等,或者上述的任意合适的组合。
[0206]
可以以一种或多种程序设计语言或其组合来编写用于执行本公开操作的计算机程序代码,程序设计语言包括面向对象的程序设计语言—诸如java、smalltalk、c++,还包括常规的过程式程序设计语言—诸如”c”语言或类似的程序设计语言。程序代码可以完全地在用户计算机上执行、部分地在用户计算机上执行、作为一个独立的软件包执行、部分在用户计算机上部分在远程计算机上执行、或者完全在远程计算机或服务器上执行。在涉及远程计算机的情形中,远程计算机可以通过任意种类的网络——包括局域网(lan)或广域网(wan)—连接到用户计算机,或者,可以连接到外部计算机(例如利用因特网服务提供商来通过因特网连接)。
[0207]
根据本发明的再一方面,还涉及一种电子设备,包括:
[0208]
一个或多个处理器;以及
[0209]
存储装置,存储一个或多个程序,
[0210]
当所述一个或多个程序被所述一个或多个处理器执行,使得所述一个或多个处理器实现如上所述的方法中的步骤ii)。
[0211]
可选的,电子设备还可以包括收发器。处理器和收发器相连,如通过总线相连。需要说明的是,实际应用中收发器不限于一个,该电子设备的结构并不构成对本技术实施例的限定。
[0212]
其中,处理器可以是cpu,通用处理器,dsp,asic,fpga或者其他可编程逻辑器件、晶体管逻辑器件、硬件部件或者其任意组合。其可以实现或执行结合本技术公开内容所描述的各种示例性的逻辑方框,模块和电路。处理器也可以是实现计算功能的组合,例如包含一个或多个微处理器组合,dsp和微处理器的组合等。
[0213]
总线可包括一通路,在上述组件之间传送信息。总线可以是pci总线或eisa总线等。总线可以分为地址总线、数据总线、控制总线等。存储器802可以是rom或可存储静态信息和指令的其他类型的静态存储设备,ram或者可存储信息和指令的其他类型的动态存储设备,也可以是eeprom、cd-rom或其他光盘存储、光碟存储(包括压缩光碟、激光碟、光碟、数字通用光碟、蓝光光碟等)、磁盘存储介质或者其他磁存储设备、或者能够用于携带或存储具有指令或数据结构形式的期望的程序代码并能够由计算机存取的任何其他介质,但不限于此。
[0214]
下面将结合实施例对本发明的实施方案进行详细描述。
[0215]
实施例1构建数据库
[0216]
本实施例以16s rdna为例说明参考序列数据库的构建过程。
[0217]
1)下载公共数据库中最新的16s rdna参考序列数据,保证数据库序列来源的完备性。下载数据库包括但不限于ncbi及silva数据库,数据库包含的参考序列种类和数量可根据具体需要选择,例如,可包含100、250、500、1000、2000、10,000种、甚至全部已有种类等等不同的数量。
[0218]
2)去除参考序列中的富集扩增引物以及两端引物之外的序列
[0219]
本步骤的目的是获取不包含引物的参考序列。可以使用基于smith-waterman局部比对算法或者其他比对定位短序列的方法,根据一定的相似性(例如但不限于80%、85%、90%、95%等)匹配富集时使用的扩增引物(本步骤中简称扩增引物)信息及在参考序列中定位富集扩增引物,然后根据引物匹配的位置进行序列剪切,去除扩增引物以及引物之外的两端序列。另外,也可以仅仅去除引物以外的两端序列。
[0220]
3)对16s rdna序列进行物种注释校正
[0221]
由于数据库中部分参考序列的物种注释信息可能存在误差,为了确保物种注释信息能够最大程度上反映其正确的物种归属,本技术方案对物种注释进行校正。但如果获取的参考序列物种注释信息足够确信,亦可不做此校正。校正流程:将每条参考序列与ncbi nt/nr库进行blast比对,按照序列相似性99.5%、覆盖度(coverage)99%(适当提高或者降低相似性值也在此技术范围内)的规则从ncbi nt/nr库中筛选出匹配的参考序列集。在匹配的参考序列集的物种注释中选择最具代表性的物种分类,并使用该分类信息校正所针对
的参考序列的物种注释。同时根据该代表性物种分类在匹配的参考序列集的物种注释中的唯一性程度对所针对的参考序列的物种注释的确定性进行分级,物种注释的确定性级别包括:certain,limitedcertainty,rare annotation(按确定性由高到低排序),也可以根据实际确定性情况进行更多/少级别的的分类,甚至是不分类。
[0222]
4)对16s rdna序列中的模糊碱基进行校正
[0223]
数据库中某些参考序列的部分位置的碱基不明确或包含简并碱基。为了明确这些位置最具代表性的碱基,我们按照规则进行校正,主要步骤是:对在同一个物种内相似性大于97%、或97.5%、或98%、或98.5%等的参考序列使用muscle进行多重序列比对,对于模糊碱基位置使用最具代表性的碱基进行替换。此步骤之后若还存在模糊碱基,则选择其同属中具有97%、或97.5%、或98%、或98.5%等相似性的参考序列进行多重序列比对,对于模糊碱基位置使用最具代表性的碱基进行替换。处理之后依然存在的模糊碱基予以保留。
[0224]
5)根据物种注释以及相似性对数据库进行去冗余
[0225]
本步骤主要是为了去除100%相似的冗余参考序列,同时确保需要的参考序列不被误去除。因此,本技术方案要求被去除的参考序列需被其他参考序列所包含、以及物种注释的确定性等级较低。
[0226]
6)对物种内非冗余的参考序列按照指定阈值的相似性进行聚类
[0227]
按照相似性98%、或98.5%、或99%、或99.5%等(相似性指标可根据实际数据库参考序列的情况进行适当调整)的标准,对每个物种内所有的非冗余参考序列进行聚类。聚类后,每个参考序列类(cluster)中的代表序列作为该类的seed,与它聚在同一个cluster里的其他序列作为其子序列。
[0228]
7)对物种间相似性较高的类进行合并后按照指定阈值的相似性重聚类
[0229]
对6)中得到的所有seed按照98%、或98.5%、或99%等(可根据6)中seed聚类的相似性指标作出适当调整)相似性进行聚类,将聚在同一个类里的不同物种的seed及其子序列合并,然后按照99.5%相似性进行重聚类,用新获得的cluster替换参与重聚类计算的旧cluster。另外,本步骤可以和6)合并,直接使用所有的序列进行聚类,而非按照前面描述的先物种内聚类、再合并物种间相似的类。但如果距离较近的物种太多,则直接做聚类的方式并不一定能达到很好的分析效果。
[0230]
8)将聚类后较大的cluster按照更高相似性进行拆分
[0231]
在cluster中seed的子序列太多的情况下,按照更高相似性(例如99.6%、99.8%、99.9%等)标准对该cluster进行拆分,用拆分后形成的新cluster替换掉拆分前的cluster。至此,构建了包含两层聚类层级结构的数据库:第一层由cluster的seed参考序列组成,第二层包含所有cluster的子参考序列。
[0232]
9)对构建的cluster的seed参考序列进行层级聚类
[0233]
依次按照97%、98%、99%(仅以此3种聚类相似性指标为代表,只要能达到逐层聚类的效果都可以)的相似性对所有seed参考序列进行聚类。主要步骤为:首先对所有的seed参考序列按照97%相似性聚类,得到第一层的cluster(包含seed及其子序列);然后针对每个cluster分别对其内部的参考序列按照98%相似性进行聚类,得到第二层的cluster;之后对第二层的每个cluster分别对其内部的参考序列按照99%相似性进行聚类,得到第三层的cluster。最终构建出包含数据库中所有seed参考序列的索引层级结构,使得每个seed
参考序列获得分别对应于三种不同序列相似性聚类的seed id。至此,所有参考序列的索引信息构成了一个由四列信息组成的文件。参考序列数据库的seed聚类层级关系如下图所示。
[0234]
实施例二:模拟实验及数据分析
[0235]
模拟数据的生成
[0236]
从数据库中人为选取单个或不同组合的多个参考序列生成模拟样本。将参考序列随机打断以生成长度分布符合正态分布的reads,随机生成指定测序深度的reads集合以模拟测序结果。通过模拟共产生了83个由不同物种等量混合构成的样本,其中包括24个单物种样本和59个多物种混合(物种数2-12不等)样本,参考序列reads的平均深度从20x到800x不等。
[0237]
分析流程
[0238]
数据库:该批次数据分析使用的是包含2119种致病细菌的数据库,同时按照97%、98%、99%相似性构建参考序列的层级关系,共包含一级代表序列34,025个,总序列数83,886条(本版本参考序列为去除冗余的参考序列数量)。
[0239]
由于本数据库seed层数量较大,接下来使用hisat2软件按照参数
--
score-min l,-1,-0.08
--
no-spliced-alignment
--
no-templatelen-adjustment
--
secondary-k 100000
--
mm
--
no-softclip进行比对,然后按照1%mismatch reads rate对alignment进行过滤,并将alignment中因pcr重复而产生的reads去除,然后统计每个参考序列比对的reads数目,将比对reads数大于10条的记为“有read比对”,否则为“无read比对”,然后在按照聚类层级关系使用富集分析的方法,按照较宽松的富集p值0.1筛选出显著富集reads的seed id,提取对应的seed作为下一步的候选参考序列。
[0240]
使用bowtie2将reads与执行步骤2)中得到的候选参考序列比对,比对参数为
--
min-score l,-1,-0.1
–
a,参考序列间不竞争reads。然后按照0.5%mismatch reads rate对alignment进行过滤,并将alignment中因pcr重复而产生的reads去除,计算每个参考序列的覆盖度并根据参数cv≤0.55&gap≤40&mean depth≥20进行过滤,通过阈值筛选的参考序列作为下一步的候选序列。
[0241]
将reads与步骤3)中筛选得到的参考序列进行比对,使用bowtie2软件,参数同3),然后按照0.5%的mismatch reads rate对alignment进行过滤,并将alignment中因pcr重复而产生的reads去除,计算每个参考序列的cv、gap、cor、cv/cor、mean depth指标,按照cv≤0.55&cor≥0.6&mean depth≥20&gap≤40的指标,并进行逐步迭代筛选,直到所有参考序列满足要求。
[0242]
对4)筛选出的参考序列进行筛选。如果覆盖度参数满足cv≤0.4&cor≥0.9&gap==0,则直接进入最终的迭代,否则需要进入参考序列的二级cluster内部进行竞争迭代筛选。二级cluster内部迭代筛选步骤同3)和4),其中同4)步骤的筛选阈值参数为cv≤0.55&cor≥0.6&mean depth≥20&gap≤25,满足条件的参考序列进入下一步分析。
[0243]
将5)筛选出的所有满足条件的参考序列合并,进行终极迭代筛选。使用bowtie2比对,参数同4),使用0.5%的mismatch reads rate对alignment进行过滤,并将alignment中因pcr重复而产生的reads去除。对于参考序列比对位点中优势碱基占比小于0.95的位点仅保留支持参考序列的reads比对,计算每个参考序列的覆盖度,根据阈值cv≤0.55&cor≥
0.6&mean depth≥20&gap≤25进行逐步迭代筛选,逐一末位淘汰进行迭代筛选。直到所有参考序列都满足阈值之后,再对筛选得到的每个物种内的参考序列两两进行多重序列比对。最终满足要求的参考序列作为最终的目标序列,对应的物种为目标物种,同时计算每个物种的reads数目以及在样本中的占比。
[0244]
分析结果
[0245]
模拟实验数据的分析表明,样本物种检测的平均灵敏度(sensitivity)达98.65%,平均精确性(precision)达98.79%,其中,单物种样本的灵敏度和精确性均为100%。
[0246]
实施例三
[0247]
本实施例以革兰氏阳性菌作为实验对象,为液体培养基培养的已知物种分类的单纯菌种。目的是考察本技术方案对样本中的微生物是否能够正确鉴定、以及考察本技术方案检测能力的灵敏度下限。
[0248]
(一)样本准备
[0249]
实验室培养表皮葡萄球菌单菌种样本,经琼脂平板培养计数,确认实验样本中加入的细菌cfu数量。投入量为2560cfu,样本编号为inq19m0101、inq19m0102。投入量为640cfu,样本编号为inq19m0103、inq19m0104。投入量为160cfu,样本编号为inq19m0105、inq19m0106。投入量为40cfu,样本编号为inq19m0107、inq19m0108。投入量为10cfu,样本编号为inq19m0109、inq19m0110。
[0250]
(二)实验过程
[0251]
1.样本dna提取,使用ezup柱式细菌基因组dna抽提试剂盒,生工生物工程(上海)股份有限公司,货号b518255-0100。按照试剂盒说明书提取样本中的dna。
[0252]
在本实施例中,选取的样本为细菌,提取方案为提取样本中包含的dna。在本技术方案中,提取的核酸也可能为rna,或者dna和rna同时提取;所使用的核酸提取试剂盒也不限于该厂家或者该货号。
[0253]
2.目的基因扩增。pcr扩增引物为:f:5'agagtttgatcmtggctcag 3'、或者5'agagtttgatcctggctcag 3'、或者5'ctcctacgggaggcagcag 3'、或者5'gtgccagcmgccgcgg 3'、或者5'aaactyaaakgaattgacgg 3'、或者5'gcaacgagcgcaaccc 3'、或者5'agagtttgatcatggctcag 3'、或者5’aactgaagagtttgatcctggctc 3’;r:5’、ggttaccttgttacgactt 3’、或者5'ggytaccttgttacgactt 3'、或者5'ctgctgcsycccgtag 3'、或者5'gwattaccgcggckgctg 3'、或者5'ccgtcaattcmtttragttt 3'、或者5'gggttgcgctcgttg 3'、或者5'aaggaggtgwtccarcc 3'、或者5'tagggttaccttgttacgactt 3'、或者5’tacggttaccttgttacgactt 3’。所用的试剂为ex taq hs,宝日医生物技术(北京)有限公司,货号rr006a。在本实施例中,所用的富集靶向序列的方法为pcr富集16s rrna基因序列。在本技术方案中,所用富集方法还可能为杂交捕获;pcr所用的酶也不局限于该公司的该货号,可能是其他货号或者其他公司的taq酶,也可能是其他类型的酶,只要能够扩增dna序列即可;对于细菌等原核生物,本实施例选取了16s rrna基因的序列作为靶向序列,但是靶向序列存在多种可能性,包括但是不限于16s/18s/its等区域的序列,根据测序目的的不同可以任意选择和组合,即使针对原核生物,靶向序列也不局限于16s,扩增引物也不局限于实施例中所列举的序列;实施例中所列举的反应体系与扩增条件,仅仅是针对
当前所用的taq酶和所用引物的适宜反应条件,不作为对本技术方案的限制。
[0254]
反应体系为:
[0255]
组分体积(μl)10*buffer7.5dntps6ex taq0.375f3r3模板量10ddh2o45.125
[0256]
反应条件为:
[0257][0258][0259]
3.产物纯化,使用的主要试剂为dna分选磁珠(无锡百迈格生物科技有限公司,bmsx)。操作步骤如下:本实施例中,使用dna分选磁珠对pcr产物进行纯化。在本技术方案中,纯化方式不局限于使用磁珠进行纯化,也可能是吸附柱纯化等其他可以纯化pcr产物的方式;磁珠纯化也不局限于dna分选磁珠,也不局限于该公司的产品,能够纯化pcr产物的磁珠均可;甚至,是否需要进行纯化,或者纯化形式的选择,要根据上一步对靶向序列的富集方式、以及下一步反应所用的试剂和反应条件进行选择,也可能不需要进行纯化,也可能不是对“pcr产物”进行纯化。
[0260]
a)将已室温平衡30min的磁珠涡旋震荡3min,充分混匀,按照相对应编号向96孔板的对应孔中加入37.5μl磁珠。
[0261]
b)待pcr反应结束后,取下pcr管,离心10s,将pcr产物转移至上步对应好编号的96孔板中,封板膜密封,涡旋震荡30s,静置5min,瞬时离心,放置磁力板上静置5min,小心吸走上清液。
[0262]
c)加入180μl新配制的75%乙醇,手持磁力板底部,将96孔板水平移动至相邻板槽中反复3-5次,静置5s,待液体澄清后吸走上清。
[0263]
d)步骤(3)重复一次。
[0264]
e)将96孔板从磁力板上取下,瞬时离心,重新放回磁力板,用100μl排枪将剩余乙醇吸走,敞开盖晾干至磁珠出现细微裂痕。
[0265]
f)加入22μl ddh2o,封板膜密封,涡旋震荡30s,静置5min,瞬时离心。将96孔板放置于磁力板上静置至澄清,将上清液全部转移至新的1.5ml低吸附离心管中,即为纯化产物。
[0266]
4.pcr产物定量,使用qubit 3.0试剂(qubit dsdna br assay kit,thermo fisher scientific q32850),对纯化好的pcr产物进行定量。按照试剂与仪器的说明书进行操作。在本实施例中,pcr产物的定量使用了荧光染料法。在本技术方案中,对于靶向富集后的核酸,定量方式不局限于荧光染料法,也可能是其他染料法或者非染料法,比如紫外分光光度计法、荧光定量pcr法、毛细管电泳或微流控电泳结合核酸染料荧光成像法等;对于荧光染料法,也不局限于该公司或者该货号的试剂;所用方法可以准确反应样本中的核酸的质量(指物质含量,并非品质)即可;如果使用紫外分光光度法,还能够获得核酸品质的信息,这些信息也属于本技术方案质量控制的范畴,不构成对本技术方案的创新。
[0267]
5.目的基因片段化。本实验使用脱氧核糖核酸酶i(dnase i)对待测核酸进行片段化处理,dnase i来自于new england biolabs,货号为m0303s。操作步骤如下:本实施例中,使用核酸酶dnase i对样本中的核酸进行打断。本技术方案中,所用的方法不局限于使用酶等生物学方法,也可能是超声等物理学方法,或者化学方法,或者其他类型的方法,能够可控的将长片段的核酸打断为短片段,同时不引入会影响核酸序列识别的其他改变即可;即使用生物学方法,也不局限于dnase i这种酶,还可能是其他能够切断dna的酶,例如其他dna内切酶、dna转座酶(如tn5转座酶)、crispr等;使用dnase i这种酶,也不局限于该公司或者该货号;反应条件仅为配合本实施例中所用的试剂,随着试剂与方法的不同,反应条件有可能改变,不构成对本技术方案的限制。在本实施例中,从上一步得到的核酸中取出5-100ng加入本反应。本技术方案中,5-100ng不是一个限制条件,仅仅是一个较为合适的条件,更多或者更少的核酸也可以用于本技术方案,0.01ng或者更少的核酸,使用本技术方案也能得到正确的结果。本技术方案中,所用的dnase i的量仅为当前条件下一个较为合适的量,并不是限制性因素。本技术方案中,纯化方式的说明同前。
[0268]
a)将样本的16s rrna纯化产物、酶缓冲液、edta及mncl2从冰箱中拿出,融化,震荡,瞬时离心,备用。
[0269]
b)取出与样本相对应数量的pcr管,放置冰盒上,加入60ng的16s rrna纯化产物xμl,同时补水(30-x)μl。edta按1:4的比例稀释配置后备用(终浓度1.25mmol/l)。
[0270]
c)原浓度酶(2u/μl)稀释为0.1u/μl:取1个0.2ml的pcr管,放置冰盒上,加入19μl1*dnaseⅰbuffer(10*buffer稀释),然后从冰箱中取出dnaseⅰ酶,瞬时离心后,取1μl的酶加入到pcr管中,混合点震荡30-40次,瞬时离心后立即放置冰盒上。
[0271]
d)配制酶、酶缓冲液以及mncl
2 mix:用0.01u酶切(30ng dna):
[0272]
i.取1个1.5ml的离心管,放置冰盒上,依次往里面加入10*dnaseⅰ缓冲液((n+2)*2)μl、mncl2((n+2)*2)μl以及稀释后的dnaseⅰ酶(0.1u/μl)((n+2)*0.1)μl,加入完成后,点震荡40-50次,瞬时离心后立即放置冰盒上。
[0273]
ii.依次取4.1μl mix加入到八连排中,然后用排枪取15μl步骤2中所配溶液按照对应顺序加入到八联排中,改好管盖,点震荡5-10次,瞬时离心后立即放置冰盒上。
[0274]
e)将八联排管放入pcr仪上,20℃15min反应。
[0275]
f)反应后,取出八连排,加入5μl稀释后的edta终止反应,加入后,震荡,瞬时离心,得到的产物为弥散片段。立即进入下一步的磁珠纯化步骤。
[0276]
6.片段化产物纯化。使用的主要试剂为dna分选磁珠(无锡百迈格生物科技有限公司,bmsx)。操作步骤如下:
[0277]
a)将已室温平衡30min的磁珠涡旋震荡3min,充分混匀,按照相对应编号向96孔板的对应孔中加入50μl磁珠。
[0278]
b)将酶切产物转移至上步对应好编号的96孔板中,封板膜密封,涡旋震荡30s,静置5min,瞬时离心,放置磁力板上静置5min,小心吸走上清液。
[0279]
c)加入180μl新配制的75%乙醇,手持磁力板底部,将96孔板水平移动至相邻板槽中反复3-5次,静置5s,待液体澄清后吸走上清。
[0280]
d)步骤(3)重复一次。
[0281]
e)将96孔板从磁力板上取下,瞬时离心,重新放回磁力板,用100μl排枪将剩余乙醇吸走,敞开盖至磁珠出现细微裂痕。
[0282]
f)加入42μl te,封板膜密封,涡旋震荡30s,静置5min,瞬时离心。将96孔板放置于磁力板上静置至澄清,吸取40.2μl上清液转移至新的96孔板中,即为纯化产物。
[0283]
7.dna末端修复,使用试剂为pfu酶(天根生化科技(北京)有限公司,ep101)、dntp mixture(天根生化科技(北京)有限公司,cd111),dna分选磁珠(无锡百迈格生物科技有限公司,bmsx)。操作步骤如下:本实施例中,末端修复后的dsdna为平末端,即dna的双链末端齐平。本技术方案中,对小片段的末端修复根据所选用的测序平台的不同而不同,例如选用illumina测序平台时,修复后的dsdna的一条链末端会有一个突出的腺嘌呤(a),修复方式不构成本技术方案的限制条件;末端修复选用的酶也不局限于pfu酶,也可能是taq酶或者其他酶类;选用pfu酶,也不局限于该公司的该货号。在本实施例中,在末端修复步骤之后,进行核酸片段长度筛选。在本技术方案中,片段筛选不局限于使用dna片段分选磁珠,也可能是其他方式,例如将核酸电泳之后选取所需长度片段的核酸并回收,能够对核酸片段进行选择即可;所使用的dna片段分选磁珠也不限于该公司或者该货号;筛选后保留的片段长度与所选用的测序仪器、测序试剂、测序参数设置有关,不构成对本技术方案的限制;对核酸片段长度的筛选,也不局限于在末端修复之后,也可能在末端修复之前,或者在下一步、下下一步实验之后,这一调整不构成对本技术方案的限制。本技术方案中,纯化方式的说明同前。
[0284]
a)在装有40μl dna的低吸附管中依次加入试剂,并使用移液器充分吹打,震荡混匀,瞬时离心,置于pcr仪上72℃反应15min。
[0285][0286][0287]
b)将反应后的dna产物加入装有32.5μl磁珠的96孔板中,封板膜密封,涡旋震荡30sec,静置5min,瞬时离心,放置磁力板上静置5min,将上清对应转入另外新的装有10μl磁
珠的96孔板中,封板膜密封,涡旋震荡30sec,静置5min,瞬时离心,放置磁力板上静置5min,小心吸走上清液。
[0288]
c)加入180μl新配制的75%乙醇,手持磁力板底部,将96孔板水平移动至相邻板槽中反复3-5次,静置5sec,待液体澄清后吸走上清。
[0289]
d)步骤(c)重复一次。
[0290]
e)将96孔板从磁力板上取下,瞬时离心,重新放回磁力板,用100μl排枪将剩余乙醇吸走,敞开盖晾干至磁珠出现细微裂痕。
[0291]
f)加入27μl te,封板膜密封,涡旋震荡30sec,静置5min,瞬时离心。将96孔板放置于磁力板上静置至澄清,吸取25μl上清液转移至新的96孔板中,即为纯化产物。
[0292]
8.测序接头连接。所用试剂为t4 dna连接酶(thermo fisher scientific,el0011),dna分选磁珠(无锡百迈格生物科技有限公司,bmsx),操作步骤如下:本实施例中,测序接头指测序引物,所用的测序引物为thermo fisher公司ion torrent测序平台的测序引物。在本技术方案中,测序引物的选择受到测序仪器、测序试剂的影响,不构成对本技术方案的限制;所用的连接酶也不局限于t4连接酶,能够将两段核酸片段连接起来的酶或者其他技术方法均可以使用;选用t4连接酶,也不局限于该公司或者该货号;反应体系的比例以及反应条件,仅为当前较为合适的条件,不构成对被技术方案的限制。本技术方案中,纯化方式的说明同前。
[0293]
a)根据下表在96孔pcr板中依次加入试剂,涡旋振荡5sec,瞬时离心,将管子置于pcr仪上,25℃反应30min。
[0294][0295][0296]
b)将反应后的dna产物加入装有40μl磁珠的96孔板中,封板膜密封,涡旋震荡30sec,静置5min,瞬时离心,放置磁力板上静置5min,小心吸走上清液。
[0297]
c)加入180μl新配制的75%乙醇,手持磁力板底部,将96孔板水平移动至相邻板槽中反复3-5次,静置5sec,待液体澄清后吸走上清。
[0298]
d)步骤(c)重复一次。
[0299]
e)将96孔板从磁力板上取下,瞬时离心,重新放回磁力板,用100μl排枪将剩余乙醇吸走,敞开盖晾干至磁珠出现细微裂痕。
fisher scientific q32850),对纯化好的pcr产物进行定量。按照试剂与仪器的说明书进行操作。在本实施例中,测序文库的定量使用了荧光染料法。在本技术方案中,对于构建完成的测序文库,定量方式不局限于荧光染料法,也可能是其他染料法或者非染料法,比如紫外分光光度计法、荧光定量pcr法、毛细管电泳或微流控电泳结合核酸染料荧光成像方法等;对于荧光染料法,也不局限于该公司或者该货号的试剂;所用方法可以准确反应样本中的核酸的质量(指物质含量,并非品质)即可;如果使用紫外分光光度法,还能够获得核酸品质的信息,这些信息也属于本技术方案质量控制的范畴,不构成对本技术方案的创新。
[0312]
11.混合测序文库。将同一批次测序的不同样本的测序文库进行混合,根据qubit定量的结果,不同样本都加入相等的量,制作混合文库。本实施例中,不同样本的测序文库为等量混合。在本技术方案中,不同样本的混合也可以是不等量;每次混合的样本数量可以根据测序仪器、测序试剂、测序方式、实验实际需求等情况灵活调整,不构成对本技术方案的限制。
[0313]
12.混合文库定量。使用ion library taqman quantitation kit(thermo fisher scientific,4468802),按照试剂说明书进行操作。本实施例中,使用荧光定量pcr对混合后的文库进行定量。在本技术方案中,定量方式也可以是其他染料法或者非染料法,例如qubit、紫外分光光度计、agilent 2100生物分析仪等;选用荧光定量pcr法,也不局限于该公司或者该货号的试剂;甚至,此步骤的定量不是必须进行的,省略此步骤也不构成对本技术方案的限制;如果使用紫外分光光度法或者agilent 2100等仪器,还能够获得核酸品质的信息,这些信息也属于本技术方案质量控制的范畴,不构成对本技术方案的创新。
[0314]
13.进行测序,使用ion one touch 2测序准备平台和ion torrent pgm测序平台,使用试剂为ion pgmhi-q view ot2 kit(thermo fisher scientific,a29900),ion pgm hi-q view sequencing kit(thermo fisher scientific,a30044),ion316 chip kit v2 bc(thermo fisher scientific,4488149)按照相应仪器与试剂说明书进行操作。在本实施例中,选取了thermo fisher的ion torrent pgm测序仪和ion 316测序芯片以及配套的测序试剂、测序方法进行实验。在本技术方案中,实验流程兼容的ngs测序仪厂家包括但不限于thermo fisher,illumina,bgi等主流厂家所有目前在市场销售的仪器与试剂以及测序方式,这些均不构成对本技术方案的限制。
[0315]
(三)数据分析
[0316]
下机数据经过数据过滤,滤除低质量的reads,剩余高质量的clean data方可用于后期分析。分析流程如下:
[0317]
1.数据库:包含252种重要临床致病微生物,包含代表序列2396个,代表序列选取与前述数据库构建流程中的一级seed选取方法类似,区别是直接对所有序列按照99.5%相似性聚类,选择每个类的seed作为代表组成了代表集。
[0318]
2.将高质量的测序reads与人类基因组序列hg19版本序列进行比对,使用bowtie2默认参数,将比对上人基因组的序列去除。
[0319]
3.直接进行代表集序列不竞争reads的比对及覆盖统计,主要方法:使用bowtie2(参数
–
a;或者将reads分别与每个参考序列比对,使用默认参数)进行比对,然后按照0.5%mismatch reads rate进行过滤,并将alignment中因pcr重复而产生的reads去除,计算每个参考序列的覆盖度并根据参数cv≤0.5&gap≤15&mean depth≥20进行过滤。
[0320]
4.将3)过滤后的参考序列按照98%相似性进行聚类,筛选出每个cluster的代表序列进入下一步分析。
[0321]
5.将reads与4)筛选得到的参考序列进行比对,使用bowtie2软件的默认参数,然后按照0.5%的mismatch reads rate对alignment进行过滤,并将alignment中因pcr重复而产生的reads去除,计算每个参考序列的cv、gap、cor、cv/cor、mean depth指标,按照cv≤0.4&cor≥0.7&mean depth≥20&gap≤15的指标进行逐步迭代过滤,,直到所有参考序列满足要求。然后计算每个物种的reads数目及其占比情况。
[0322]
(四)实验结果
[0323]
10个样本的平均检测序列数量为55,931条,在所有样本中,表皮葡萄球菌都可以正确检出,灵敏度为100%。
[0324][0325][0326]
实施例四
[0327]
本实施例以革兰氏阴性菌作为实验对象,为使用液体培养基培养的已知物种分类的单纯菌种。目的是考察本技术方案对样本中微生物是否能够正确鉴定,以及考察本技术方案检测能力的灵敏度下限。
[0328]
(一)样本准备
[0329]
单菌种样本(粘质沙雷菌),使用琼脂平板培养计数,确认加入的细菌cfu数。投入量为2560cfu,样本编号为inq19m0111-inq19m0112。投入量为640cfu,样本编号为inq19m0113-inq19m0114。投入量为160cfu,样本编号为inq19m0115-inq19m0116。投入量为40cfu,样本编号为inq19m0117-inq19m0118。投入量为10cfu,样本编号为inq19m0119-inq19m0120。
[0330]
(二)实验过程
[0331]
同实施例三。
[0332]
(三)数据分析
[0333]
同实施例三。
[0334]
(四)实验结果
[0335]
10个样本的平均检测序列数量为54,703条,除样本inq19m0119外,其余样本中粘质沙雷菌都可以正确检出。
[0336][0337]
实施例五
[0338]
使用液体培养基培养的已知物种分类的单纯菌种,考察本技术方案是否可以正确鉴定多个微生物物种混合的样本。
[0339]
(一)样本准备
[0340]
对5种微生物(金黄色葡萄球菌、表皮葡萄球菌、粘质沙雷菌、绿脓杆菌、大肠埃希菌)使用琼脂平板培养计数,确认细菌cfu数。等量混合所有菌株(200cfu),样本编号为inq19m0133、inq19m0134。
[0341]
(二)实验过程
[0342]
同实施例三。
[0343]
(三)数据分析
[0344]
同实施例三。
[0345]
(四)实验结果
[0346]
2个样本的平均检测序列数量为62,970条,两个样本中,加入的5个物种都可以正确检出。
[0347][0348]
实施例六
[0349]
使用液体培养基培养的已知物种分类的单纯菌种,本实施例的目的是考察本技术方案能否正确鉴定含有多个物种的样本中占比很低的物种。
[0350]
(一)样本准备
[0351]
2种实验室培养的微生物菌株(表皮葡萄球菌、粘质沙雷菌)用琼脂平板培养计数,确认细菌cfu数。按16倍稀释梯度混合(粘质沙雷菌混合量3200cfu,表皮葡萄球菌混合量200cfu)构成样本,样本编号为inq19m0143、inq19m0144。
[0352]
(二)实验过程
[0353]
同实施例三。
[0354]
(三)分析方法
[0355]
同实施例三。
[0356]
(四)实验结果
[0357]
2个样本的平均检测序列数量为46,026条,两个样本中,含量相差16倍的两个物种都能够正确检出。
[0358][0359]
实施例七
[0360]
考察本技术方案检测临床样本的能力。
[0361]
(一)样本准备
[0362]
来自医院采集的用于病原微生物鉴定的胆汁样本,编号inq19m0322。
[0363]
(二)实验过程
[0364]
同实施例三。
[0365]
(三)数据分析
[0366]
1.数据库:包含252种重要临床致病微生物的数据库,包含一级代表序列1920个,总序列数143009条(本版本参考序列未去冗余)。
[0367]
2.将高质量的测序reads与人类基因组序列hg19版本序列进行比对,使用bowtie2默认参数,将比对上人基因组的序列去除。
[0368]
3.直接将reads与一级seed参考序列进行不竞争reads的比对、覆盖统计及筛选。主要方法为:使用bowtie2(参数
--
min-score l,-0.6,-0.15-a)进行序列比对,再按照0.5%mismatch reads rate对alignment进行过滤,并将alignment中因pcr重复而产生的reads去除,然后计算每个参考序列的覆盖度并根据参数cv≤0.5&gap≤25&mean depth≥20进行过滤,通过阈值的参考序列作为下一步的候选。
[0369]
4.将3、过滤后的参考序列按照98%相似性进行聚类,筛选出每个cluster的代表序列seed进入下一步分析。
[0370]
5.进行参考序列间竞争reads的比对并根据覆盖情况逐步迭代筛选出满足要求的参考序列。具体步骤是:将reads与4、筛选得到的参考序列进行比对,使用bowtie2软件的默认参数,然后按照0.5%的mismatch reads rate对alignment进行过滤,并将alignment中因pcr重复而产生的reads去除。对于参考序列比对位点中优势碱基占比小于0.95的位点仅保留支持参考序列的reads比对。计算每个参考序列的cv、gap、cor、cv/cor、mean depth指标,然后按照阈值cv≤0.5&cor≥0.65&mean depth≥20&gap≤25的指标进行逐步迭代筛选,直到所有参考序列满足要求。满足要求的参考序列作为下一步的候选。
[0371]
6.将步骤5筛选出的候选序列进行seed内部迭代筛选。主要步骤是:首先对这些seed按照覆盖情况进行分类,满足阈值cv≤0.4&cor≥0.9&gap==0的参考序列直接进入最终迭代筛选;不满足的参考序列,分别对其进行seed内部的迭代筛选,筛选方法与3、4、两步一样,其中同步骤3的迭代筛选参数为cv≤0.5&gap≤15&mean depth≥20,同步骤4的迭代筛选参数为cv≤0.5&cor≥0.7&mean depth≥20&gap≤25,将所有满足要求的候选seed合并,作为终极候选参考序列。
[0372]
7.将reads与步骤6得到的终极候选参考序列比对,按照0.5%的mismatch reads rate对alignment进行过滤,并将alignment中因pcr重复而产生的reads去除。计算每个参考序列的cv、gap、cor、cv/cor、mean depth指标,然后按照阈值cv≤0.5&cor≥0.7&mean depth≥20&gap≤25进行逐步迭代筛选。所有满足要求的seed作为最终的目标序列,对应的物种为检测的物种,同时计算每个物种的read count以及在样本中的占比。
[0373]
(四)实验结果
[0374]
样本的检测序列数量为55,115条,人源序列占所有检测序列的0.020%。本技术的检测结果显示样本中含有大肠埃希菌、屎肠球菌,此结果与使用微生物培养等方法得到的结果一致。
[0375][0376]
实施例八
[0377]
考察本技术方案检测临床样本的能力。
[0378]
(一)样本准备
[0379]
来自医院采集的用于病原微生物鉴定的胸水样本,编号inq19m0324。
[0380]
(二)实验过程
[0381]
同实施例三。
[0382]
(三)数据分析
[0383]
同实施例七。
[0384]
(四)实验结果
[0385]
样本的检测序列数量为67,797条,人源序列占所有检测序列的9.50%。本技术的检测结果显示,样本中含有大肠埃希菌,此结果与使用微生物培养等方法得到的结果一致。
[0386][0387]
实施例九
[0388]
比较本技术方案与当前最常用的微生物鉴定方法的检测能力,对比指标包括检测阳性率、阳性结果一致性,考察本技术方案检测临床样本的能力。
[0389]
(一)样本准备
[0390]
来自医院采集的用于病原微生物鉴定的一批样本,收集时间2019.08-2019.08,有效样本共89例。样本采集后分为两份,分别使用本技术方案或者细菌培养的方式进行鉴定。样本类型包括:胆汁、胸腹腔积液、关节腔积液、脑脊液、尿液、脓、心包积液、引流液、痰等,囊括了临床微生物检验的主要样本类型。
[0391]
(二)实验过程
[0392]
同实施例三。
[0393]
(三)数据分析
[0394]
1.数据库:包含1.8万多个物种(含所有已知的细菌、支原体、衣原体、立克次体、螺旋体)。按照97%、98%、99%相似性构建参考序列的层级关系,共包含一级代表序列30,816个,总序列数154,392条(本版本参考序列为去除冗余的参考序列数量)。
[0395]
2.将高质量的测序reads与人类基因组参考序列hg38版本进行比对,使用bowtie2默认参数,将比对上人基因组的序列去除。
[0396]
3.使用hisat2软件按照参数
--
score-min l,-1,-0.08
--
no-spliced-alignment
--
no-templatelen-adjustment
--
secondary-k 100000
--
mm
--
no-softclip进行比对,然后按照1%mismatch reads rate对alignment进行过滤,并将alignment中因pcr重复而产生的reads去除。统计每个参考序列比对的reads数目,将比对reads数大于10条的记为“有read比对”,否则为“无read比对”。按聚类层级关系,根据富集分析的方法,按照较宽松的富集p值0.1筛选出显著富集reads的seed id,提取对应的seed作为下一步的候选参考序列。
[0397]
4.使用bowtie2将reads与步骤3得到的候选参考序列比对,比对参数为
--
min-score l,-1,-0.1
–
a,参考序列间不竞争reads。然后按照0.5%mismatch reads rate对alignment进行过滤,并将alignment中因pcr重复而产生的reads去除。计算每个参考序列的覆盖度并根据参数cv≤0.6&gap≤50&mean depth≥20进行过滤,通过阈值的参考序列作为下一步的候选序列。
[0398]
5.将reads与步骤4筛选得到的参考序列进行比对,使用bowtie2软件,参数同步骤4。然后按照0.5%的mismatch reads rate对alignment进行过滤,并将alignment中因pcr重复而产生的reads去除。对于参考序列比对位点中优势碱基占比小于0.95的位点仅保留支持参考序列的reads比对。计算每个参考序列的cv、gap、cor、mean depth指标,按照cv≤0.6&cor≥0.65&mean depth≥20&gap≤25的指标,并进行逐步迭代筛选,直到所有参考序列满足要求。
[0399]
6.如果覆盖度参数满足cv≤0.4&cor≥0.9&gap==0,则步骤5筛选出的参考序列直接进入最终的迭代,否则,需要进入参考序列的二级cluster内部进行竞争迭代筛选,二级cluster内部迭代筛选步骤同4、和5,筛选的阈值参数为cv≤0.6&cor≥0.7&mean depth≥20&gap≤25。
[0400]
7.将步骤6筛选出的所有满足条件的参考序列合并,进行终极迭代筛选,使用bowtie2比对,参数同步骤4。然后使用0.5%的mismatch reads rate对alignment进行过滤,并将alignment中因pcr重复而产生的reads去除。计算每个参考序列的覆盖度,根据阈值cv≤0.6&cor≥0.7&mean depth≥20&gap≤25进行逐步迭代筛选。最终满足要求的参考序列作为最终的目标序列,对应的物种为目标物种,同时计算每个物种的reads数目以及在样本中的占比。
[0401]
(四)实验结果
[0402]
全部样本使用本技术方案的检测阳性率为98.9%(88/89)。在使用细菌培养鉴定方法检测出的阳性样本中,本技术方案与细菌培养鉴定检测结果的符合度为90.6%(48/53)。
[0403]
[0404]
[0405]
[0406]
[0407]
[0408]
[0409]
[0410]
[0411]
[0412]
[0413]
[0414]
[0415]
[0416]
[0417][0418]
实施例十
[0419]
本实施例用于说明本技术方案中,在某一环节加入一定量已知序列的核酸之后,可以提高对检测结果中潜在的污染结果鉴别的能力。
[0420]
(一)样本准备
[0421]
来自医院采集的用于病原微生物鉴定的引流液样本,样本采集后分为两份,分别使用本技术方案或者细菌培养的方式进行鉴定。
[0422]
(二)实验流程
[0423]
同实施例三。增加部分包括:步骤2,目的基因扩增时,在配制pcr反应的反应液中加入一定量的含有已知序列的核酸的质粒载体,样本提取时的阴性对照得到的核酸提取产物也使用加入了相同量的已知序列的核酸,同时进行反应。加入的核酸序列长度与16s rrna基因的序列长度近似。本实施例选择在此步骤加入已知序列的核酸。在本技术方案中,加入已知序列的核酸并不是必须的,不加入并不会影响本技术方案的检测能力;已知序列的核酸不局限于在此步骤加入,也可以在核酸提取前加入,或者在本步骤之后加入,或者在本技术方案任意认为合适的步骤加入;加入的核酸序列可以单独存在,也可以使用质粒、病毒、细胞等作为载体;加入的核酸序列量可以从1个copy至无限多个copy的范围。
[0424]
(三)分析流程
[0425]
1.数据库:包含2202个临床致病菌物种。同时按照97%、98%、99%相似性构建参考序列的层级关系,共包含一级代表序列40,035个,总序列数92,385条(参考序列为去除冗余的参考序列数量)。
[0426]
2.使用hisat2软件按照参数
--
score-min l,-1,-0.08
--
no-spliced-alignment
--
no-templatelen-adjustment
--
secondary-k 100000
--
mm
--
no-softclip进行比对,然后按照1%mismatch reads rate对alignment进行过滤,并将alignment中因pcr重复而产生的reads去除。然后统计每个参考序列比对的reads数目,将比对reads数大于10条的记为“有read比对”,否则为“无read比对”。按聚类层级关系根据富集分析的方法,按照较宽松的富集p值0.1筛选出显著富集reads的seed id,提取对应的seed作为下一步的候选参
考序列。
[0427]
3.使用bowtie2将reads与2、得到的候选参考序列比对,比对参数为
--
min-score l,-1,-0.08
–
a,参考序列间不竞争reads,然后按照1%mismatch reads rate对alignment进行过滤,去除alignment中由于pcr导致的重复序列。计算每个参考序列的覆盖度并根据参数cv≤0.6&end gap≤40&middle gap≤10&mean depth≥20进行过滤,通过阈值的参考序列作为下一步的候选序列。
[0428]
4.将3、筛选得到的参考序列利用排列组合的方式进行随机分组,然后对每组进行比对迭代筛选。将reads分别与每组筛选得到的参考序列进行比对,使用bowtie2软件,参数同3、。按照1%的mismatch readsrate对alignment进行过滤,去除alignment中由于pcr导致的重复序列。计算每个参考序列的nrmse、end gap、middle gap、cor、mean depth等指标,按照nrmse≤0.35&cor≥0.6&mean depth≥20&end gap≤40&middle gap≤10的指标,进行逐步迭代筛选,直到所有参考序列满足要求。
[0429]
5.将4、筛选出的所有参考序列,分别进入参考序列的二级cluster内部进行竞争迭代筛选。二级cluster内部迭代筛选步骤同3、和4、,但是筛选的阈值更严格一些:比对参数为
--
min-score l,-1,-0.04
–
a;两步的reads mismatch rate仅允许0.5%;同步骤3、的筛选参数为end gap≤25&middle gap《1&cv≤0.55&meandepth≥20;同步骤4、的参数为nrmse≤0.3&cor≥0.6&mean depth≥20&end gap≤25&middle gap《1。满足条件的参考序列进入下一步分析。
[0430]
6.将5、筛选出的所有满足条件的参考序列合并,进行终极迭代筛选。使用bowtie2比对,参数同5、,使用0.5%的mismatch reads rate对alignment进行过滤,并将alignment中因pcr重复而产生的reads去除,对于参考序列比对位点中优势碱基占比小于0.95的位点仅保留支持参考序列的reads比对。计算每个参考序列的覆盖度,根据阈值为nrmse≤0.3&cor≥0.65&mean depth≥20&end gap≤25&middle gap《1进行逐步迭代筛选,最终得到满足要求的参考序列作为最终的目标序列,对应的物种为目标物种,同时计算每个物种的reads数目以及在样本中的占比。
[0431]
7.对上述筛选得到的参考序列进行背景污染筛选。一是如果参考序列不在重要临床致病物种列表且占比小于5%的物种直接去除;二是对于检测到内标序列的样本中的物种,如果这些物种在对照样本中也有检测到,计算临床样本中该物种与内标的比值与所有对照样本中该物种与内标比值符合同一个正态分布的概率。如果概率大于0.05,则认为该物种是背景污染并从检测结果中去除,否则予以保留。然后重新计算所有物种在样本中的占比,得到最终的分析结果。
[0432]
(四)实验结果
[0433]
使用本技术方案对原始样本进行检测,原始检测结果中包括已知序列的核酸(inq internal standard)(相对含量1)、疮疱丙酸杆菌(相对含量0.064)、粪肠球菌(相对含量0.5781)。通过与同批次的阴性对照样本的检测结果对比,以与已知序列核酸的相对含量为计算依据,可以判定疮疱丙酸杆菌为污染,最终报告结果为粪肠球菌,与使用微生物培养等方法得到的结果一致。
[0434][0435]
实施例和对比例的产品的表征数据以及效果数据
[0436]
本发明实施例三至实施例八中的全部样本,平均每个样本的测序数据为55,663条短序列数据,有效数据量在本实施例中为90%以上,对靶序列的覆盖度接近100%。
[0437]
实施例七中,样本的检测序列数量为55,115条短序列,人源序列占所有检测序列的0.020%。共有7,707条短序列比对至enterococcus faecium(屎肠球菌)物种,占比13.98%,物种目标基因序列测序覆盖率99.9%,平均测序深度349.04
×
。共有2,761条短序列比对至escherichia coli(大肠埃希菌)物种,占比5.01%,物种目标基因序列测序覆盖率99.8%,平均测序深度140.85
×
。
[0438]
实施例八中,样本的检测序列数量为67,797条,人源序列占所有检测序列的9.50%。共有3,136条短序列比对至escherichia coli(大肠埃希菌)物种,占比4.63%,物种目标基因序列测序覆盖率100%,平均测序深度397.54
×
。
[0439]
作为对比,new england journal of medicine,2014,370(25):2408-2417的案例报道中,采用宏基因组方法(mngs)检测患者样本。样本的测序数据为8,187,737条短序列,其中98.7%的数据为人类基因组序列,总共有475条短序列比对至leptospiraceae科,在总数据量中占比为0.0006%(万分之0.6),物种基因组测序覆盖率2.2%-3.7%,基因组平均测序深度为0.02-0.04
×
。案例对比显示,mngs技术检测微生物能够获得的有效数据量、有效数据占比、以及对目标基因组序列的覆盖度及测序深度均远不及本技术方案。
[0440]
本发明实施例九中,89个临床样本用于检测。本技术方案的检测阳性率为98.9%
(88/89),高于其他非ngs方法(细菌培养结合质谱鉴定方法)的阳性率(59.6%,53/89)。在非本技术方案检测阳性的样本中,本技术方案检测结果与前者的一致率为90.6%(48/53)。
[0441]
同时,与另一案例进行比较,clinical infectious diseases(2018)67(s2):s231
–
40的报道中,采用宏基因组方法检测患者样本的阳性率为38.2%(195/511),同组样本非ngs方法的阳性率为27.0%(138/511)。在使用非mngs方案检测阳性的样本中,mngs方案检测结果与前者的一致率为60.9%(84/138)。由此可见,本技术方案在临床样本检测中具有比现有技术更高的阳性率。同时,与相关技术mngs相比,本技术方案具有更高的传统细菌培养检测阳性一致率。
[0442]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0443]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。