1.本发明涉及有机合成技术领域,尤其涉及一种三萜衍生物及其制备方法和应用。
背景技术:
2.艾滋病是由人类免疫缺陷病毒(hiv)感染而引起的一种重大传染性疾病,迄今已夺走了近3300万人的生命。目前临床治疗艾滋病大多采用高效抗逆转录病毒疗法,该疗法通过合并3个或多个抗hiv药物来治疗艾滋病,以减少其单一抗药性,可有效降低病毒载量,同时可以显著降低感染艾滋病后的死亡率。该方法的出现和应用将hiv感染从一种致死性疾病转变为可控、可治的慢性传染性疾病。但是,由于病毒的高突变率和基因重组会导致多个耐药菌株的出现,耐药性不断发生,使其临床治疗效果大大降低。此外,使用该疗法无法根除hiv病毒库,患者需终身服用药物,长期使用药物造成的毒副作用及各种临床并发症的发生都限制了该疗法的应用。
3.天然药物是人类预防和治疗疾病的重要物质来源,由于其结构的多样性、作用的有效性、毒副作用低以及来源广泛等特点,受到国内外学者的广泛重视。从天然资源中寻找新的抗hiv药物和先导化合物是目前国内外新药研发的重要研究方向。据文献报道,从狭叶南五味子、大叶黄栀子以及鹧鸪麻等植物中分离得到了一系列具有抗hiv活性的天然环菠萝蜜烷型三萜皂苷(wu hf,morris-natschke sl,xu xd,et al.recent advances in natural anti-hiv triterpenoids and analogs.med res rev,2020,40:2339-2385.);从中药黄三七中分离得到的四环三萜化合物铁破锣皂苷i具有明显的抗hiv活性,运用有机合成的方法对该化合物的c-3位进行结构修饰,获得抗艾滋病活性显著提高的衍生物(20s,24s)-15β,16β-diacetoxy-18,24;20,24-diepoxy-9,19-cyclolanostane-3β,25-diol 3-o-3',3'-dimethylsuccinate(wu hf,ma gx,yang qw,et al.discovery and synthesis of novel beesioside i derivatives with potent anti-hiv activity.eur j med chem,2019,166:159-166.)。
技术实现要素:
4.鉴于此,本发明的目的在于提供一种四环三萜衍生物及其制备方法和应用。本发明提供的四环三萜衍生物可用于抗艾滋病毒药物的制备,具有明确的选择性抗艾滋病毒活性。
5.为了实现上述发明目的,本发明提供以下技术方案:
6.一种四环三萜衍生物,具有式i所示的结构:
[0007][0008]
本发明还提供了上述技术方案所述的四环三萜衍生物的制备方法,包括以下步骤:
[0009]
将四环三萜皂苷化合物铁破锣皂苷i进行酶水解,得到铁破锣皂苷i的苷元;
[0010]
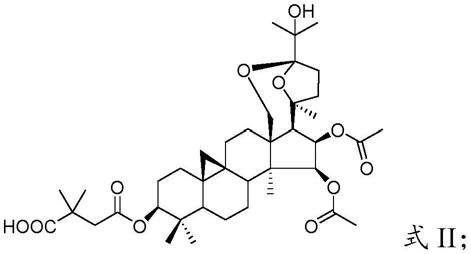
将所述铁破锣皂苷i的苷元与2,2-二甲基琥珀酸酐反应,得到具有式ii所示结构的化合物;
[0011][0012]
将所述具有式ii所示结构的化合物与氢氧化钾乙醇溶液混合,进行第一去乙酰基反应,得到所述具有式i中结构1所示的四环三萜衍生物;
[0013]
将所述具有式ii所示结构的化合物与溴苄混合,对羧基进行酯化反应,得到所述具有式i中结构2所示的四环三萜衍生物;
[0014]
将所述具有式i中结构2所示的四环三萜衍生物与氢氧化钾乙醇溶液混合进行第二次乙酰基反应,得到具有式i中结构3所示的四环三萜衍生物。
[0015]
优选地,所述第一去乙酰基反应和第二去乙酰基反应的温度独立地为-10~10℃,时间独立地为0.5~3h。
[0016]
优选地,所述羧基酯化反应的温度为50~100℃,时间为3~24h。
[0017]
优选地,所述铁破锣皂苷i的苷元与2,2-二甲基琥珀酸酐反应在微波条件下进行,所述微波条件的温度为120~160℃,时间为1~3h。
[0018]
本发明还提供了上述技术方案所述的四环三萜衍生物或上述技术方案所述制备方法制得的四环三萜衍生物在制备抗艾滋病药物中的应用。
[0019]
优选地,所述抗艾滋病药物包含有效剂量的所述四环三萜衍生物、其立体异构体、可药用盐和药学上可接受的载体、辅料、赋形剂和稀释剂。
[0020]
优选地,所述抗艾滋病药物的剂型包括片剂、注射剂、胶囊剂、颗粒剂、丸剂、散剂、口服液、缓释制剂、控释制剂或纳米制剂药学上可接受的剂型。
[0021]
优选地,当所述四环三萜衍生物具有式i中结构1所示时,具有体外抗hiv-1作用,在hiv-1
nl4-3
病毒感染的mt-4细胞中,对hiv病毒具有抑制作用。
[0022]
本发明提供了具有式i所示结构的四环三萜衍生物,可用于抗艾滋病毒药物的制备,具有明确的选择性抗艾滋病毒活性。
具体实施方式
[0023]
本发明提供了一种四环三萜衍生物,具有式i所示的结构:
[0024][0025]
在本发明中,当r1和r2同时为h时,所述具有式i中结构1所示的四环三萜衍生物的化学名称为:(20s,24s)-18,24;20,24-diepoxy-9,19-cyclolanostane-3β,15β,16β,25-tetraol-3-o-3
′
,3
′‑
dimethylsuccinate,当r1为bn,r2为ac时,所述具有式i中结构2所示的四环三萜衍生物的化学名称为:(20s,24s)-15β,16β-diacetoxy-18,24;20,24-diepoxy-9,19-cyclolanostane-3β,25-di ol-3-o-4
′‑
(benzyloxy)carbonyl-3
′
,3
′‑
dimethylsuccinate,当r1为bn,r2为h时,所述具有式i中结构2所示的四环三萜衍生物的化学名称为:(20s,24s)-18,24;20,24-diepoxy-9,19-cyclolanostane-3β,15β,16β,25-tetraol-3-o-4
′‑
(benzyloxy)carbonyl-3
′
,3
′‑
dimethylsuccinate。
[0026]
本发明还提供了上述技术方案所述的四环三萜衍生物的制备方法,包括以下步骤:
[0027]
将四环三萜皂苷化合物铁破锣皂苷i进行酶解,得到铁破锣皂苷i的苷元;
[0028]
将所述铁破锣皂苷i的苷元与2,2-二甲基琥珀酸酐反应,得到具有式ii所示结构的化合物;
[0029][0030]
将所述具有式ii所示结构的化合物与氢氧化钾乙醇溶液混合进行第一去乙酰基反应,得到所述具有式i中结构1所示的三萜衍生物;
[0031]
将所述具有式ii所示结构的化合物与溴苄混合进行苄基保护反应,得到所述具有式i中结构2所示的三萜衍生物;
[0032]
将所述具有式i中结构2所示的三萜衍生物与氢氧化钾乙醇溶液混合进行第二去乙酰基反应,得到具有式i中结构3所示的三萜衍生物。
[0033]
本发明将三萜皂苷类化合物铁破锣皂苷i进行酶解,得到铁破锣皂苷i的苷元。
[0034]
在本发明中,所述酶解优选在水解酶molsin和磷酸氢二钠-柠檬酸缓冲液混合进行
[0035]
本发明对所述三萜皂苷类化合物铁破锣皂苷i的来源没有特殊的限定,采用本领域技术人员熟知的制备方法制得即可。在本发明的实施例中,所述四环三萜皂苷化合物铁破锣皂苷i优选通过以下步骤得到:
[0036]
取黄三七souliea vaginata(maxim.)franch.10kg,干燥粉碎后,采用不同倍量、不同比例乙醇/水、甲醇/水或者丙酮/水回流或冷浸提取,减压回收溶剂得提取物220g,提取物溶解于水里,依次经过石油醚、三氯甲烷、乙酸乙酯和正丁醇萃取,乙酸乙酯萃取部位35g经硅胶(100~200目)柱层析,石油醚/乙酸乙酯(100:0~1:1)、二氯甲烷/甲醇(50:1~1:1)洗脱,获得极性不同的流份,取中等极性的流份8g,经硅胶柱层析(200~300目)石油醚/乙酸乙酯(10:1~1:1)、二氯甲烷/甲醇(20:1~5:1)洗脱,并经反相柱层析甲醇/水(50:50~100:0)洗脱,并经凝胶lh-20甲醇洗脱得活性前体化合物粗品,最后经甲醇重结晶得到三萜皂苷类化合物铁破锣皂苷i。
[0037]
在本发明中,所述四环三萜皂苷化合物铁破锣皂苷i与水解酶molsin的质量比优选为1:1~1:10。
[0038]
在本发明中,所述磷酸氢二钠-柠檬酸缓冲液的ph值优选为4.0。
[0039]
在本发明中,所述混合优选在无水乙醇中进行。在本发明中,所述混合优选先将四环三萜皂苷化合物铁破锣皂苷i用无水乙醇溶解,向该溶液中加入溶解于纯水的水解酶molsin(aspergillus saitoi)和0.2m磷酸氢二钠-0.1m柠檬酸缓冲液(ph 4.0)。
[0040]
在本发明中,所述反应的温度优选为37℃,所述反应的时间优选为2天。
[0041]
反应结束后,本发明优选将所得反应液用等体积的乙酸乙酯萃取3次,乙酸乙酯部分合并,经无水硫酸钠干燥、浓缩后,经硅胶柱(200~300目)层析,用正己烷/丙酮(10:1~1:1)洗脱,并经甲醇重结晶,得到铁破锣皂苷i的苷元。
[0042]
在本发明中,所述铁破锣皂苷i的苷元的结构如式iii所示:
[0043][0044]
得到铁破锣皂苷i的苷元后,本发明将所述铁破锣皂苷i的苷元与2,2-二甲基琥珀酸酐反应,得到具有式ii所示结构的化合物。
[0045]
在本发明的实施例中,所述具有式ii所示结构的化合物优选通过以下步骤得到:
[0046]
取四环三萜皂苷化合物铁破锣皂苷i酶解后的苷元,将苷元与2,2-二甲基琥珀酸酐在无水吡啶和4-二甲氨基吡啶(dmap)中进行微波合成反应。
[0047]
在本发明中,所述苷元与2,2-二甲基琥珀酸酐的摩尔比优选为1:1~1:10,反应温
度优选为150~160℃,反应时间优选为1~3h。
[0048]
所述微波合成反应完成后,将所得反应液加入1n盐酸中和,然后加入乙酸乙酯萃取3次,乙酸乙酯部分用brine洗涤3次,加入无水硫酸镁干燥,经硅胶柱(200~300目)层析,正己烷/丙酮梯度洗脱(体积比10:1~1:1),得到具有式ii所示结构的化合物。
[0049]
得到所述具有式ii所示结构的化合物,本发明将所述具有式ii所示结构的化合物与氢氧化钾乙醇溶液混合进行第一去乙酰基反应,得到所述具有式i中结构1所示的三萜衍生物。
[0050]
在本发明中,所述氢氧化钾优选以koh乙醇溶液的形式加入,所述koh乙醇溶液的质量分数优选为2.5%。
[0051]
在本发明中,所述第一去乙酰基反应的反应温度优选为-10~10℃,反应时间优选为0.5~3h。
[0052]
第一去乙酰基反应结束后,本发明优选在反应液中加入与koh等摩尔量的盐酸中和以终止反应,用等体积的乙酸乙酯萃取3次,乙酸乙酯部分合并,经无水硫酸钠干燥、浓缩后,经硅胶柱(200~300目)层析,用石油醚/丙酮(体积比10:1~1:1)洗脱,得到式i中结构1所示的四环三萜衍生物。
[0053]
得到所述具有式ii所示结构的化合物,本发明将所述具有式ii所示结构的化合物与溴苄混合进行苄基保护反应,得到所述具有式i中结构2所示的三萜衍生物。
[0054]
在本发明中,所述苄基保护反应优选采用dmf作为反应溶剂。
[0055]
在本发明中,所述苄基保护反应优选在碱性环境中进行,本发明优选利用碳酸钾提供碱性环境。
[0056]
在本发明中,所述具有式ii所示结构的化合物、溴苄与碳酸钾的摩尔比优选为1:1:2。
[0057]
在本发明中,所述苄基保护反应的温度优选为50~100℃,反应时间优选为3~24h。
[0058]
苄基保护反应结束后,本发明优选在所得反应液中加入等体积水,用等体积的乙酸乙酯萃取3次,乙酸乙酯部分合并,经无水硫酸钠干燥、浓缩后,经硅胶柱(200~300目)层析,用石油醚/丙酮(体积比10:1~1:1)洗脱,得到式i中结构2所示的三萜衍生物。
[0059]
得到所述具有式i中结构2所示的三萜衍生物后,本发明将所述具有式i中结构2所示的三萜衍生物与氢氧化钾混合进行第二去乙酰基反应,得到具有式i中结构3所示的三萜衍生物。
[0060]
在本发明中,所述氢氧化钾优选以koh乙醇溶液的形式加入,所述koh乙醇溶液的质量分数优选为2.5%。
[0061]
在本发明中,所述第二去乙酰基反应的反应温度优选为-10~10℃,反应时间优选为0.5~3h。
[0062]
第二去乙酰基反应结束后,本发明优选在反应液中加入与koh等摩尔量的盐酸中和以终止反应,用等体积的乙酸乙酯萃取3次,乙酸乙酯部分合并,经无水硫酸钠干燥、浓缩用0.22μm微膜过滤后,采用高效型制备液相色谱法,以60~95vol%甲醇洗脱进行分离纯化,得到式i中结构3所示的三萜衍生物。
[0063]
在本发明中,所述四环三萜衍生物的制备原理如下式所示:
[0064][0065]
本发明还提供了上述技术方案所述四环三萜衍生物在制备抗艾滋病药物中的应用。
[0066]
在本发明中,所述抗艾滋病药物优选包含有效剂量的所述四环三萜衍生物、其立体异构体、可药用盐和药学上可接受的载体、辅料、赋形剂和稀释剂。
[0067]
在本发明中,所述抗艾滋病药物的剂型优选包括片剂、注射剂、胶囊剂、颗粒剂、丸剂、散剂、口服液、缓释制剂、控释制剂或纳米制剂药学上可接受的剂型。
[0068]
在本发明中,当所述四环三萜衍生物具有式i中结构1所示时,具有体外抗hiv-1作用,在hiv-1
nl4-3
病毒感染的mt-4细胞中,对hiv病毒具有抑制作用。
[0069]
下面结合实施例对本发明提供的具有式i所示结构的四环三萜衍生物及其制备方法和应用进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
[0070]
实施例1
[0071]
四环三萜衍生物具有式i中结构1所示的制备方法:
[0072]
四环三萜皂苷类化合物铁破锣皂苷i的制备
[0073]
取黄三七souliea vaginata(maxim.)franch.药材10kg,干燥粉碎后,采用不同倍量、不同比例乙醇/水、甲醇/水或者丙酮/水回流或冷浸提取,减压回收溶剂得提取物220g,提取物溶解于水里,依次经过石油醚、三氯甲烷、乙酸乙酯和正丁醇萃取,乙酸乙酯萃取部位35g经硅胶(100~200目,200g)柱层析,石油醚/乙酸乙酯(100:0~1:1)、二氯甲烷/甲醇(50:1~1:1)洗脱,获得极性不同的流份,取中等极性的流份8g,经硅胶柱层析(200~300目,150g)石油醚/乙酸乙酯(10:1~1:1)、二氯甲烷/甲醇(20:1~5:1)洗脱,并经反相柱层析甲醇/水(50:50~100:0)洗脱,并经凝胶lh-20甲醇洗脱得活性前体化合物粗品,最后经甲醇重结晶得单体化合物1.1g,经核磁共振波谱和质谱,并与参考文献(n.sakurai,m.nagai,t.goto,t.inoue,p.g.xiao,studies on the constituents ofbeesia calthaefolia and souliea vaginata.iv.1)铁破锣皂苷i,a cyclolanostanol xyloside from the rhizomes of beesia calthaefolia,chem.pharm.bull.,1993,41,272-275)比对,确定为四环三萜皂苷类化合物铁破锣皂苷i。
[0074]
制备铁破锣皂苷i的苷元
[0075]
将铁破锣皂苷i(1.1g,1.53mmol)用100ml无水乙醇溶解,向该溶液中加入溶解于100ml纯水的水解酶molsin(aspergillus saitoi)2.2g和0.2m磷酸氢二钠-0.1m柠檬酸缓
冲液(ph 4.0)1000ml,此溶液体系在37℃下搅拌反应2天。反应液用等体积的乙酸乙酯萃取3次,乙酸乙酯部分合并,经无水硫酸钠干燥、浓缩后,经硅胶柱(200~300目)层析,用正己烷/丙酮(10:1~1:1)洗脱,并经甲醇重结晶,得到单体化合物铁破锣皂苷i的苷元。
[0076]
式ii所示结构的化合物的制备:
[0077]
将铁破锣皂苷i的苷元与2,2-二甲基琥珀酸酐在无水吡啶和4-二甲氨基吡啶(dmap)中进行微波合成反应,苷元与2,2-二甲基琥珀酸酐的摩尔比为1:1,反应温度为155℃,反应时间为2h。微波反应完成后将反应液加入1n盐酸中和,然后加入乙酸乙酯萃取3次,乙酸乙酯部分用brine洗涤3次,加入无水硫酸镁干燥,经硅胶柱(200目)层析,正己烷/丙酮梯度洗脱(体积比10:1),得到具有式ii所示结构的化合物。
[0078]
制备化合物1~3
[0079]
1)化合物1的制备
[0080]
在5ml 0.25%koh的乙醇溶液中,加入30mg(0.04mmol)如式ii所示的化合物,在-5℃下进行去乙酰基反应2h。反应停止后,在反应液中加入与koh等量的1n盐酸中和以终止反应,用等体积的乙酸乙酯萃取3次,将乙酸乙酯部分合并,经无水硫酸钠干燥、浓缩后,经硅胶柱(200~300目)层析,用石油醚/丙酮(体积比10:1~1:1)洗脱,得到(20s,24s)-18,24;20,24-diepoxy-9,19-cyclolanostane-3β,15β,16β,25-tetraol-3-o-3
′
,3
′‑
dimethylsuccinate(1,得率36.8%)。
[0081]
化合物1的结构确定
[0082]
化合物1,无色晶体,esims:m/z 655[m+na]
+
;1h nmr(600mhz,pyridine-d5)δ
h 0.29(1h,d,j=4.0hz,h-19),0.52(1h,d,j=4.0hz,h-19),0.69(1h,q,j=12.0hz,h-6a),0.95(3h,s,h-28),0.98(3h,s,h-29),1.04(1h,m,h-7a),1.15(1h,m,h-1a),1.25(1h,m,h-7b,11a),1.26(2h,m,h-12),1.28(1h,m,h-5),1.31(3h,s,h-30),1.48(3h,s,h-27),1.49(1h,m,h-1b,6b),1.55(6h,s,2
×
ch
3-3
′
),1.59(3h,s,h-21),1.64(1h,m,h-2a),1.66(3h,s,h-26),1.72(1h,m,h-22a),1.85(1h,m,h-8),1.92(1h,m,h-2b),2.02(1h,d,j=10.8hz,h-17),2.04(1h,m,h-11b),2.05(1h,m,h-23a),2.06(1h,m,h-22b),2.59(1h,m,h-23b),2.60(1h,d,j=15.6hz,h-2
′
a),2.96(1h,d,j=15.6hz,h-2
′
b),4.29(1h,d,j=13.2hz,h-18a),4.44(1h,d,j=13.2hz,h-18b),4.62(1h,dd,j=10.8,8.0hz,h-16),4.71(1h,d,j=8.0hz,h-15),4.85(1h,dd,j=11.4,4.2hz,h-3);
13
c nmr(pyridine-d5,150mhz)δ
c 14.0(c-30),15.9(c-29),20.7(c-9),21.2(c-6),25.3(c-21),25.4(c-26),25.9(c-27),26.3(c-28),26.6(3
′‑2×
ch3),26.8(c-7),27.0(c-11),27.6(c-2,10),29.1(c-23),30.4(c-12),30.7(c-19),32.2(c-1),40.0(c-4,22),41.2(c-3
′
),45.6(c-2
′
),47.7(c-5),48.6(c-8),51.4(c-13),52.2(c-14),52.4(c-17),65.4(c-18),72.2(c-25),80.9(c-3),83.8(c-20),84.0(c-16),84.1(c-15),112.1(c-24),171.9(c-1
′
),179.7(c-4
′
);hresi-ms:655.3822[m+na]
+
(cacld.655.3822).
[0083]
.2)化合物2的制备
[0084]
在3ml dmf中,加入30mg如式ii所示的三萜衍生物(0.04mmol),加入等当量的溴苄和两倍当量的碳酸钾,在60℃下反应8h。反应结束后,在所得反应液中加入等体积水,用等体积的乙酸乙酯萃取3次,乙酸乙酯部分合并,经无水硫酸钠干燥、浓缩后,经硅胶柱(200~300目)层析,用石油醚/丙酮(10:1~1:1)洗脱,得到(20s,24s)-15β,16β-diacetoxy-18,
24;20,24-diepoxy-9,19-cyclolanostane-3β,25-di ol-3-o-4
′‑
(benzyloxy)carbonyl-3
′
,3
′‑
dimethylsuccinate(2,得率45.2%)。
[0085]
化合物2的结构确定
[0086]
化合物2,无色晶体,esims:m/z 829[m+na]
+
;1h nmr(600mhz,pyridine-d5)δ
h 0.18(1h,d,j=4.2hz,h-19),0.50(1h,d,j=4.2hz,h-19),0.57(1h,q,j=12.0hz,h-6a),0.93(3h,s,h-28),0.95(3h,s,h-29),1.11(1h,m,h-7a),1.12(1h,m,h-1a),1.16(1h,m,h-11a),1.19(3h,s,h-30),1.23(1h,m,h-5),1.29(3h,s,h-21),1.31(1h,m,h-6b,7b),1.40(6h,s,2
×
ch
3-3
′
),1.47(1h,m,h-1b),1.54(1h,m,h-12a),1.57(3h,s,h-27),1.59(1h,m,h-8),1.65(1h,m,h-2a),1.68(3h,s,h-26),1.85(1h,m,h-2b),1.96(1h,m,h-11b),1.97(1h,m,h-22a),2.08(1h,m,h-23a),2.13(3h,s,coch3),2.15(3h,s,coch3),2.73(1h,d,j=12.0hz,h-17),2.80(1h,m,h-23b),2.82(1h,d,j=15.6hz,h-2
′
a),2.86(1h,d,j=15.6hz,h-2
′
b),2.96(1h,m,h-12b),2.99(1h,m,h-22b),4.51(1h,d,j=13.2hz,h-18a),4.58(1h,d,j=13.2hz,h-18b),4.82(1h,dd,j=12.0,4.2hz,h-3),5.35(2h,s,h-1
″
),5.69(1h,d,j=9.0hz,h-15),5.96(1h,dd,j=12.0,9.0hz,h-16),7.34(1h,m,h-5
″
),7.41(1h,t,j=7.8hz,h-4
″
,6
″
),7.50(1h,d,j=7.8hz,h-3
″
,7
″
);
13
c nmr(pyridine-d5,150mhz)δc15.8(c-29,30),19.5(c-9),20.6(c-6),21.6(2
×
coch3),25.9(3
′‑2×
ch3),26.0(c-26,27),26.2(c-28),26.5(c-7,11),27.5(c-2),27.6(c-10),28.2(c-12),31.2(c-23),31.6(c-19),32.1(c-1),32.7(c-21),38.6(c-22),39.9(c-4),41.2(c-3
′
),45.0(c-2
′
),46.0(c-13),47.0(c-5),47.3(c-8),51.8(c-14),56.4(c-17),66.7(c-18),66.9(c-1
″
),73.0(c-25),75.4(c-16),80.9(c-3),82.3(c-15),87.1(c-20),114.6(c-24),128.6(c-3
″
,7
″
),128.8(c-5
″
),129.3(c-4
″
,6
″
),137.4(c-2
″
),171.0(coch3),171.3(coch3),171.6(c-1
′
),176.9(c-4
′
);hresi-ms:829.4503[m+na]
+
(cacld.829.4503).
[0087]
3)化合物3的制备
[0088]
在3ml 0.25%koh的乙醇溶液中,加入10mg如式i所示的化合物2,在-5℃下进行水解反应2h。反应结束后,在反应液中加入与koh等量的盐酸中和以终止反应,用等体积的乙酸乙酯萃取3次,乙酸乙酯部分合并,经无水硫酸钠干燥、浓缩,用0.22μm微膜过滤后,采用高效型制备液相色谱法,以90%甲醇洗脱,在保留时间为21min时分离得到(20s,24s)-18,24;20,24-diepoxy-9,19-cyclolanostane-3β,15β,16β,25-tetraol-3-o-4
′‑
(benzyloxy)carbonyl-3
′
,3
′‑
dimethylsuccinate(3,得率53.5%)
[0089]
化合物3的结构确定
[0090]
化合物3,无色晶体,esims:m/z 745[m+na]
+
;1h nmr(600mhz,pyridine-d5)δ
h 0.29(1h,d,j=4.2hz,h-19),0.52(1h,d,j=4.2hz,h-19),0.70(1h,q,j=12.0hz,h-6a),0.90(3h,s,h-28),0.94(3h,s,h-29),1.06(1h,m,h-7a),1.16(1h,m,h-1a),1.24(1h,m,h-11a),1.28(2h,m,h-12),1.29(1h,m,h-5),1.30(1h,m,h-7b),1.31(3h,s,h-30),1.38(6h,s,2
×
ch
3-3
′
),1.47(3h,s,h-27),1.50(1h,m,h-6b),1.53(1h,m,h-1b),1.58(3h,s,h-21),1.63(1h,m,h-2a),1.66(3h,s,h-26),1.72(1h,m,h-22a),1.81(1h,m,h-8),1.90(1h,m,h-2b),2.00(1h,m,h-22b),2.03(1h,m,h-11b),2.04(1h,d,j=10.8hz,h-17),2.05(1h,m,h-23a),2.59(1h,m,h-23b),2.78(1h,d,j=16.2hz,h-2
′
a),2.81(1h,d,j=16.2hz,h-2
′
b),4.29(1h,d,j=12.0hz,h-18a),4.45(1h,d,j=12.0hz,h-18b),4.62(1h,dd,j=10.8,
8.0hz,h-16),4.70(1h,d,j=8.0hz,h-15),4.81(1h,dd,j=11.4,4.2hz,h-3),5.34(2h,s,h-1
″
),7.32(1h,m,h-5
″
),7.39(1h,t,j=7.2hz,h-4
″
,6
″
),7.48(1h,d,j=7.8hz,h-3
″
,7
″
).
13
c nmr(pyridine-d5,150mhz)δ
c 14.0(c-30),15.8(c-29),20.8(c-9),21.2(c-6),25.4(c-21,26),25.8(c-27),25.9(3
′‑2×
ch3),26.2(c-28),26.5(c-11),26.8(c-7),27.5(c-2),27.6(c-10),29.1(c-23),30.4(c-12),30.7(c-19),32.1(c-1),40.0(c-4,22),41.2(c-3
′
),45.0(c-2
′
),47.7(c-5),48.6(c-8),51.4(c-13),52.2(c-14),52.5(c-17),65.5(c-18),66.7(c-1
″
),72.2(c-25),81.3(c-3),83.8(c-20),84.0(c-16),84.1(c-15),112.1(c-24),128.6(c-3
″
,7
″
),128.7(c-5
″
),129.3(c-4
″
,6
″
),137.4(c-2
″
),171.6(c-1
′
),176.9(c-4
′
);hresi-ms:745.4289[m+na]
+
(cacld.745.4292).
[0091]
式i化合物在hiv-1
nl4-3
感染的mt-4细胞中抑制hiv活性试验
[0092]
待测试活性的药物:
[0093]
(20s,24s)-18,24;20,24-diepoxy-9,19-cyclolanostane-3β,15β,16β,25-tetraol-3-o-3
′
,3
′‑
dimethylsuccinate(1)
[0094]
(20s,24s)-15β,16β-diacetoxy-18,24;20,24-diepoxy-9,19-cyclolanostane-3β,25-diol-3-o-4
′‑
(benzyloxy)carbonyl-3
′
,3
′‑
dimethylsuccinate(2)
[0095]
(20s,24s)-18,24;20,24-diepoxy-9,19-cyclolanostane-3β,15β,16β,25-tetraol-3-o-4
′‑
(benzyloxy)carbonyl-3
′
,3
′‑
dimethylsuccinate(3)
[0096]
测试方法:
[0097]
样品对hiv-1抑制作用的体外评价试验参照文献进行z.dang,l.zhu,w.lai,h.bogerd,k.h.lee,l.huang,c.h.chen,aloperine and its derivatives as a new class ofhiv-1entry inhibitors.acs med.chem.lett.7(2016)240-244.hiv-1nl4-3 nanoluc-sec病毒感染的mt4细胞培养于9孔板中,加入不同浓度的待测化合物,hiv-1
nl4-3 nanoluc-sec病毒是一种报告病毒,具有secnluc作为报告基因,化合物用dmso溶解后,采用promega nano-glo luciferase assay system通过检测荧光素激酶的活性来检测病毒的复制。测定结果见表1。由表1可以看出,(20s,24s)-15β,16β-diacetoxy-18,24;20,24-diepoxy-9,19-cyclolanostane-3β,25-diol-3-o-4
′‑
(benzyloxy)carbonyl-3
′
,3
′‑
dimethylsuccinate(2)被认为是最有效的抗hiv物质,其半数有效抑制hiv浓度ec
50
值为0.28μm,cc
50
值为22.1μm,且治疗指数ti值大于78,有可能发展成为天然来源的抗hiv-1药物。
[0098]
表1样品及阳性药物在mt-4细胞上抑制hiv-1活性试验结果a
[0099][0100]a采用多周期病毒复制的方法测试对hiv-1病毒的抑制.
[0101]
b ec
50
:半数有效抑制hiv浓度(mean+/-sd of3 tests).
[0102]
c cc
50
:半数有效抑制细胞浓度
[0103]dti:cc
50
/ec
50
.
[0104]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。