1.本发明属于无机分子筛技术合成领域,涉及高效耐酸性丝光沸石分子筛的制备方法。
背景技术:
2.丝光沸石(mor)是工业上重要的一类微孔分子筛,具有均匀的孔道结构、适宜的耐酸性以及良好的水热稳定性,具有沿c轴方向的平行的椭圆形十二元环(0.65
×
0.70nm)和八元环(0.26
×
0.57nm)直孔道,且两个平行孔道通过b轴方向的八元环孔道(0.34
×
0.48nm)相连通。丝光沸石作为催化材料广泛应用于石油化工领域,在二甲醚羰基化、烷基化、加氢裂解等反应过程中表现出优异的催化性能,由于其良好的耐酸性和水热稳定性,通过在撑体上制备丝光沸石膜也可用于苛刻条件下比如乙酸/水等混合体系中的酸的纯化或酯化反应的耦合,提高反应产率。
3.目前,合成丝光沸石的方法一般分为有机模板剂导向法和无模板剂法。尽管有机模板剂价格昂贵并且后续模板剂去除的过程极易造成环境污染,还是有多数研究人员通过模板剂来合成丝光沸石,且还停留在实验室水平。文献[advanced powder technology 23(2012)757
–
760]中报道了通过调节凝胶组成和结晶条件的情况下来制备丝光沸石但是其合成时间长达6天,水热处理时间过长。专利[cn1257831a]报道了在无有机模板剂的情况下,使用廉价的水玻璃作为硅源,在晶化了54h后得到了结晶度相对较高的丝光沸石晶体。专利[cn 110790283 a]同样公开报道了在不使用有机模板剂的情况下,使用超细颗粒硅酸(颗粒直径小于1微米)作为硅源合成硅铝比较高的丝光沸石的方法,但是所需水热合成晶化时间长达5天,耐酸性未知。
技术实现要素:
[0004]
针对以上合成丝光沸石利用有机模板剂污染环境,合成时间长效率低、以及长期强耐酸性未知的问题,本发明提供一种高效耐酸性丝光沸石分子筛的制备方法,采用在合成母液中同时加入氟源和晶种,大幅缩短了所需的时间,且制备出的丝光沸石具有长期耐酸性,并使得其在极短的合成时间内结晶强度提升。此外,本发明提供的快速制备耐酸性丝光沸石的方法无模板剂,合成方法简单,合成时间短,是一种绿色、高效、节约资源的制备方法,有利于大规模工业生产。
[0005]
为了达到上述效果,本发明采用的技术方案为:
[0006]
一种高效耐酸性丝光沸石分子筛的制备方法,包括如下步骤:
[0007]
(1)将硅源、铝源、碱源按照一定的比例混合,在室温下搅拌(搅拌时间优选为1-3h)并老化为溶胶状;
[0008]
(2)再加入氟源,搅拌后得到合成母液(搅拌时间优选为10-30min),(进一步的控制合成母液中各组分的摩尔比为(6-10)na2o:(30-40)sio2:(1.2-1.8)al2o3:(1200-1800)h2o:(6-15)f-)。
[0009]
(3)向合成母液中加入丝光沸石晶种,搅拌使其分散均匀(搅拌时间可以为10-15min)。作为优选,控制晶种质量浓度为0.3-1.5wt%,晶种粒径在2-10μm。
[0010]
(4)将步骤(2)制备的含有晶种的母液加入高压反应釜中,晶化反应结束后,用去离子水离心清洗至中性,干燥。
[0011]
进一步的,步骤(1)中碱源为氢氧化钠、氨水中的任意一种或两者的组合。
[0012]
进一步的,步骤(1)中硅源为为硅溶胶、二氧化硅及正硅酸乙酯中的任意一种或多种。
[0013]
进一步的,步骤(1)中铝源为偏铝酸钠、氧化铝中的任意一种或两者的组合。
[0014]
进一步的,步骤(1)中氟源为naf、kf、nh4f中的任意一种或多种。
[0015]
进一步的,步骤(4)中,晶化温度为160-200℃,晶化时间为6-20h。
[0016]
与现有技术相比,本发明取得了如下有益效果:
[0017]
(1)在合成沸石之前,在合成母液中加入氟源作为矿化剂和微结构优化剂能够调节铝元素在分子筛中的空间分布,使铝元素分布均匀,达到耐酸的效果,同时和加入晶种,都具有诱导成核的双重作用,大幅缩短了以往合成丝光沸石所需的高温晶体生长时间,该方法合成的丝光沸石具有耐酸的优点,该法合成简单,合成时间短,对环境友好,便于操作,适合工业化生产。
[0018]
(2)经x射线衍射仪(xrd)和场发射扫描电镜(sem)分析,本发明在极大降低合成时间后,仍能形成高结晶度的丝光沸石,大小为微米级(2-3μm),经过1mol/l的hcl溶液处理后的结果表明其在酸处理之后表面形貌保持良好,结晶强度减少较小。
附图说明
[0019]
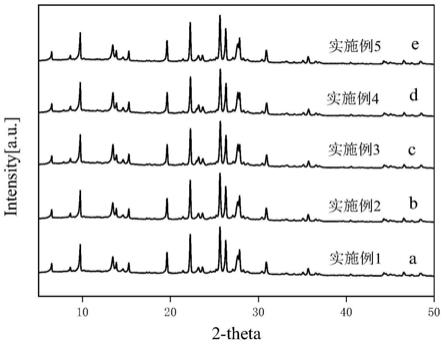
图1为按实施例1(图1中a)、实施例2(图1中b)、实施例3(图1中c)、实施例4(图1中d)、实施例5(图1中e)合成的丝光沸石分子筛的x射线粉末衍射谱图(xrd);
[0020]
图2为按实施例1(图2中a)、实施例2(图2中b)、实施例3(图2中c)、实施例4(图2中d)、实施例5(图2中e)合成的丝光沸石分子筛酸处理后的x射线粉末衍射谱图(xrd);
[0021]
图3为按对比例1(图3中a)、对比例2(图3中b)、对比例3(图3中c)、对比例4(图3中d)、对比例5(图3中e)、对比例6(图3中f)、对比例7(图3中g)合成的丝光沸石分子筛的x射线粉末衍射谱图(xrd);
[0022]
图4为按对比例4(图4中a)、对比例5(图4中c)合成的丝光沸石分子筛的xrd图,按对比例4(图4中b)、对比例5(图4中d)酸处理之后的丝光沸石分子筛的xrd图;
[0023]
图5为按实施例1(图5中(a))、实施例2(图5中(c))、实施例3(图5中(e))、实施例4(图5中(g))、实施例5(图5中(i))合成的丝光沸石分子筛的扫描电镜图(sem);实施例1酸处理后(图5中(b))、实施例2酸处理后(图5中(d))、实施例3酸处理后(图5中(f))、实施例4酸处理后(图5中(h))及实施例5酸处理后(图5中(j))的丝光沸石分子筛的扫描电镜图(sem);
[0024]
图6为按对比例1(图6中(a))、对比例2(图6中(b))、对比例3(图6中(c))、对比例6(图6中(d))的晶化产物的sem图;
[0025]
图7为按对比例4(图7中(a))、对比例5(图7中(c))合成的丝光沸石分子筛的扫描电镜图(sem);对比例4酸处理后(图7中(b))、对比例5(图7中(d)酸处理后的扫描电镜图(sem);
[0026]
图8为按对比例7合成的丝光沸石的扫描电镜图(sem);
[0027]
图9实施例1的能谱面扫图(eds-mapping);
[0028]
图10为实施例2的能谱面扫图(eds-mapping);
[0029]
图11为实施例3能谱面扫图(eds-mapping);
[0030]
图12为实施例4的能谱面扫图(eds-mapping);
[0031]
图13为实施例5能谱面扫图(eds-mapping);
[0032]
图14为对比例4能谱面扫图(eds-mapping);
[0033]
图15为对比例5-能谱面扫图(eds-mapping)。
具体实施方式
[0034]
本发明不局限于下列具体实施方式,本领域一般技术人员根据本发明公开的内容,可以采用其他多种具体实施方式实施本发明的,或者凡是采用本发明的设计结构和思路,做简单变化或更改的,都落入本发明的保护范围。需要说明的是,在不冲突的情况下,本发明中的实施例及实施例中的特征可以相互组合。
[0035]
本发明下面结合实施例作进一步详述:
[0036]
实施例1
[0037]
合成液的配比为8na2o:36sio2:1.4al2o3:1600h2o:8.2naf。具体操作为:将去离子水分成三份,氢氧化钠加到一份去离子水中溶解,把硅溶胶加入到氢氧化钠溶液中,形成硅源前驱液;将偏铝酸钠加入到另一份水中溶解,形成铝源前驱液;在搅拌下把铝源前驱液逐滴滴加到硅源前驱液中,室温搅拌1h;将氟化钠溶解于最后一份去离子水中,加入到前述的溶液中,室温搅拌1h。再加入晶种并控制晶种质量浓度为0.7wt%,搅拌30min后转移至反应釜中,于180℃合成12h,晶化产物经反复离心洗涤至ph至6.5-7.5。然后在120℃干燥6h。固体产物经xrd表征为纯丝光沸石(图1a),sem表征结果为微米级晶粒,形貌呈针尖状,晶粒尺寸约2-3微米(图5a)。经eds分析(图9),al元素分布均匀。将所得丝光沸石进行酸处理,操作步骤为:浸泡在1mol/l的hcl溶液中12天,浸泡结束后,反复离心清洗至ph=6.5-7.5,120℃下干燥6h。干燥结束后,做xrd与sem分析,酸处理产物经xrd表征(图2a)与未酸处理(图1a)比较,结晶强度仅下降了3%,sem表征结果显示表面形貌保持良好(图5b)。
[0038]
实施例2
[0039]
合成配比不变,将水热合成时间改为18h,其余合成条件与实施例1完全相同,固体产物经xrd分析为纯丝光沸石(图1b),sem表征结果为微米级晶粒,形貌呈扁平状,晶粒尺寸约2-3微米(图5c),经eds分析(图10),al元素分布均匀。将所得丝光沸石进行酸处理,处理条件与实例1完全相同,酸处理产物经xrd表征(图2b)与未酸处理(图1b)比较,结晶强度仅下降了4%,sem表征结果显示表面形貌保持良好(图5d)。
[0040]
实施例3
[0041]
将合成液的配比改为9.5na2o:39sio2:1.7al2o3:1700h2o:14naf,其余合成条件与实施例1完全相同,固体产物经xrd分析为纯丝光沸石(图1c),sem表征结果为微米级晶粒,形貌呈椭圆状,晶粒尺寸约2-3微米(图5e),经eds分析(图11),al元素分布均匀。将所得丝光沸石进行酸处理,处理条件与实例1完全相同,酸处理产物经xrd表征(图2c)与未酸处理(图1c)比较,结晶强度仅下降了4%,sem表征结果显示表面形貌保持良好(图5f)。
[0042]
实施例4
[0043]
将合成液的配比改为6.5na2o:31sio2:1.3al2o3:1300h2o:7naf,其余合成条件与实施例1完全相同,固体产物经xrd分析为纯丝光沸石(图1d),sem表征结果为微米级晶粒,形貌呈椭圆状,晶粒尺寸约1-2微米(图5g),经eds分析(图12),al元素分布均匀。将所得丝光沸石进行酸处理,处理条件与实例1完全相同,酸处理产物经xrd表征(图2d)与未酸处理(图1d)比较,结晶强度仅下降了4%,sem表征结果显示表面形貌保持良好(图5h)。
[0044]
实施例5
[0045]
合成液的配比与实施例1完全相同,将加入晶种的质量浓度改为1.4wt%的晶种,其余合成条件与实施例1完全相同,固体产物经xrd分析为纯丝光沸石(图1e),sem表征结果为微米级晶粒,形貌为扁平状,晶粒尺寸约1-2微米(图5i),经eds分析(图13),al元素分布均匀。将所得丝光沸石进行酸处理,处理条件与实例1完全相同,酸处理产物经xrd表征(图2e)与未酸处理(图1e)比较,结晶强度仅下降了3%,sem表征结果显示表面形貌保持良好(图5j)。
[0046]
对比例1
[0047]
将合成液的配比改为4na2o:28sio2:1.0al2o3:1000h2o:5naf,其他制备条件和过程与实施例1相同,晶化产物经xrd分析为无定形产物(图3a),无明显的丝光沸石特征峰,sem表征结果为其中含有大量无定形产物,结果表明未合成出丝光沸石(图6a)。
[0048]
对比例2
[0049]
将合成液的配比改为11na2o:42sio2:1.5al2o3:2000h2o:20naf,其他制备条件和过程与实施例1相同,晶化产物经xrd分析为无定形产物(图3b),sem表征结果为其中含有大量无定形产物(图6b)。
[0050]
对比例3
[0051]
合成液的配比为8na2o:35sio2:1.4al2o3:1600h2o:0naf,同时不加入晶种,水热合成36h,其他制备条件和过程与实施例1相同,晶化产物经xrd表征(图3c),丝光沸石特征峰不明显,其含有较多无定型产物,sem表征结果为结晶出的丝光沸石较少(图6c)。
[0052]
对比例4
[0053]
合成液的配比为8na2o:36sio2:1.4al2o3:1600h2o:0naf,同时不加入晶种,水热合成48h,其他制备条件和过程与实施例1相同,晶化产物经xrd分析为纯丝光沸石(图3d),sem表征结果为微米级晶粒,晶粒大小为5-8μm(图7a)。经eds分析(图14),al元素分布不均,晶界有富铝现象。所得丝光沸石进行酸处理,处理条件与实例1完全相同,酸处理产物经xrd分析(图4b)结晶度下降了60%,sem表征结果(图7b)显示表面粗糙程度较大,表面有脱落现象。
[0054]
对比例5
[0055]
合成液的配比为8na2o:36sio2:1.4al2o3:1600h2o:0naf,同时加入丝光沸石晶种,晶化时间24h,其他制备条件和过程与实施例1相同,固体产物经xrd分析为纯丝光沸石(图3e),sem表征结果为微米级晶粒,晶粒尺寸约2-3微米(图7c),经eds分析(图15),al元素分布不均,晶界有富铝现象。酸处理产物经xrd分析(图4d)结晶度下降了50%,sem表征结果(图7d)显示表面粗糙程度较大,表面有脱落现象。
[0056]
对比例6
[0057]
合成液的配比与实施例1完全相同,再加入质量浓度为0.1wt%的晶种,其余合成条件与实施例1完全相同,固体产物经xrd分析为丝光沸石特征峰强度较低(图3f),sem表征结果为结晶出少量丝光沸石,其中含有大量无定形产物(图6d)。
[0058]
对比例7
[0059]
合成配比不变,将水热合成时间改为24h,其余合成条件与实施例1完全相同,固体产物经xrd分析为纯丝光沸石(图3g)相对于实施例1峰强度下降,sem表征结果(图8)为微米级晶粒晶粒,大小1-2μm,相对于实施例1合成的晶粒有溶解现象。
[0060]
结合上述实施例和对比例,在加入了晶种和氟源的条件下,缩短了晶化时间,在较短时间内合成出了结晶强度较高,良好耐酸性的丝光沸石。氟源的加入使得不需要昂贵且对环境不友好的模板剂,有利于工业生产。因为铝原子在mor晶体边缘分布丰富,而在mor晶体中心分布较少,而氟源作为矿化剂和丝光沸石的微结构优化剂,优化了丝光沸石中铝元素的分布,氯元素均匀的分布不会造成富铝晶界,提高了丝光沸石的耐酸性。
[0061]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。