一种高效构建sgrnas质粒文库的方法
技术领域
1.本发明涉及基因编辑技术,更具体的涉及一种高效构建sgrnas质粒文库的方法。
背景技术:
2.基因编辑技术能够让人类对目标基因进行定点“编辑”,从而实现对生物体基因组特定dna片段的修饰。crispr-cas系统是原核生物的一种获得性免疫系统,运用重复间隔序列转录所得的向导rna(grnas)有助于cas蛋白识别并切割外源致病rna。crispr-cas9系统是目前在真核生物中使用较为广泛的基因编辑工具,能够准确高效的实现对靶向基因的编辑。与此同时,crispr-cas9系统也被广泛应用于高通量药物靶点的筛选中。在高通量筛选应用中,针对多个靶向基因设计导向rna序列(sgrnas),运用酶切连接的方式将其插入表达载体后,形成sgrnas质粒文库。该文库经慢病毒转染的方式成功整合到表达有cas9的宿主细胞后,即可在宿主细胞内进行基因编辑,从而形成靶向基因功能缺失的细胞文库,可用于后续的药物靶点筛选。在该过程中,快速高效的构建sgrnas质粒文库作为高通量筛选的基石至关重要。
3.目前,构建sgrnas质粒文库的方法有化学转化构建法与电击转化构建方法。其中化学转化构建法因转化效率低会给sgrnas质粒文库构建带来巨大的工作量,且sgrnas质粒文库的质量有限,从其sgrnas丢失率、基因丢失率以及sgrnas整体分布等衡量,sgrnas质粒文库质量均有待提升。而电击转化构建法虽理论上可以弥补化学转化构建法所面临的不足,但电击转化构建中实验条件的调控对于电击转化效率均有较大的影响。
技术实现要素:
4.了解决现有技术存在上述问题,本发明提供了一种高效构建sgrnas质粒文库的方法。该方法包括:1. 一种高效构建sgrnas质粒文库的方法,包括:1)合成和扩增sgrna序列;2)将扩增的sgrna序列通过酶切连接,插入质粒载体;3)将步骤2)获得的包含sgrna序列的质粒载体通过电击转化感受态细胞;和4)提取质粒。
5.2. 根据项1所述的方法,其中在步骤2)中,所述质粒载体与所述sgrna的比例为1-10:1、2-9:1、3-8:1、4-7:1或5-6:1。
6.3. 根据项2所述的方法,其中所述质粒载体与所述sgrna的比例为4:1、5:1、6:1、7:1或8:1。
7.4. 根据项1所述的方法,其中在步骤3)中,其中所述质粒载体的浓度为30 ng/
µ
l-180 ng/
µ
l、40 ng/
µ
l-170 ng/
µ
l、50 ng/
µ
l-160 ng/
µ
l、60 ng/
µ
l-160 ng/
µ
l、70 ng/
µ
l-160 ng/
µ
l、80 ng/
µ
l-150 ng/
µ
l、90 ng/
µ
l-150 ng/
µ
l、100 ng/
µ
l-140 ng/
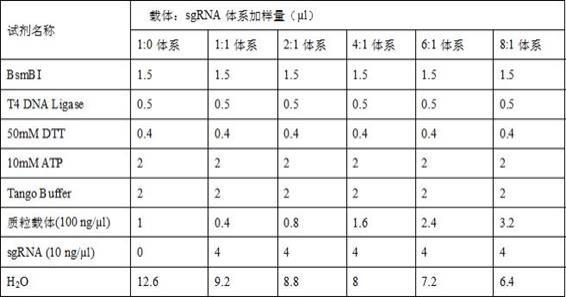
µ
l。
8.5.根据项4所述的方法,其中所述质粒载体的浓度为90 ng/
µ
l-150 ng/
µ
l。
9.6. 根据项中1-5任一项所述的方法,其中所述电击转化的电压为大于1.0kv且小于2.0kv。
10.7.根据项6所述的方法,其中所述电击转化的电压为1.4kv-1.8kv。
11.8. 根据项1-7中任一项所述方法制备的sgrnas质粒文库。
12.9. 包含项8的质粒文库的组合物。
13.10. 项9所述的组合物,其为质粒文库的储存液。
14.本发明对电击转化建库过程中涉及的关键参数进行交叉组合,通过评估电击转化效率以及文库质量等,显著降低文库构建时的工作量,为sgrna质粒文库构建提供了稳定高效的方法。
附图说明
15.图1显示合成sgrna序列的结构图;图2显示纯化浓缩后产物电转反应(1.0kv)克隆数;图3显示纯化浓缩后产物电转反应(1.4kv)克隆数;图4显示纯化浓缩后产物电转反应(1.8kv)克隆数;图5 显示泛素化酶相关基因质粒文库的sgrna分布图。
具体实施方式
16.下面结合实施例进一步说明本发明,应当理解,实施例仅用于进一步说明和阐释本发明,并非用于限制本发明。
17.除非另外定义,本说明书中有关技术的和科学的术语与本领域内的技术人员所通常理解的意思相同。虽然在实验或实际应用中可以应用与此间所述相似或相同的方法和材料,本文还是在下文中对材料和方法做了描述。在相冲突的情况下,以本说明书包括其中定义为准,另外,材料、方法和例子仅供说明,而不具限制性。以下结合具体实施例对本发明作进一步的说明,但不用来限制本发明的范围。
18.本发明提供一种高效构建sgrnas质粒文库的方法,包括:1)合成和扩增sgrna序列;2)将扩增的sgrna序列通过酶切连接,插入质粒载体;3)将步骤2)获得的包含sgrna序列的质粒载体通过电击转化感受态细胞;和4)提取质粒。
19.其中,sgrna(sgrnas)也称为单分子向导rna,是crispr基因敲除敲入系统中重要的组成部分,作用于动质体(kinetoplastid)体内一种称为rna编辑(rna editing)的后转录修饰过程中。也是一种小型非编码rna。可与pre-mrna配对,并在其中插入一些尿嘧啶(u),产生具有作用的mrna。单分子向导rna编辑的rna分子,长度大约是60~80个核苷酸,是由单独的基因转录的,具有3'寡聚u的尾巴,中间有一段与被编辑mrna精确互补的序列,5'端是一个锚定序列,它同非编辑的mrna序列互补。
20.电击转化(electrotransformation),也有人称作高压电穿孔法(high-voltage electroproration,简称电穿孔法electroproration),可用于将dna导入真核细胞(如动物细胞和植物细胞)和原核细胞(转化大肠杆菌和其他细菌等)。电转化技术的主要优点在于:
对于磷酸钙dna共沉淀法以及其他技术难以进行转化的细胞,电穿孔法仍可适用。电转化的效率高,既可用于克隆化基因的瞬间表达,也可用于建立整合有外源基因的细胞系。
21.sgrna序列的合成和扩增可以采用现有技术中已知的方法。在一个具体的实施方式中,sgrna序列的合成如图1所示,合成后的sgrna序列包含扩增用引物序列、酶切位点序列以及sgrna扩增模板序列。任何与被编辑的靶向序列互补的、适用于crispr-cas9体系的sgrna序列均可用于该体系,sgrna长度范围可以为15 nt-50 nt。
22.酶切连接包括酶切和连接两个反应,首先对扩增的sgrna序列进行纯化,纯化后的产物在限制性内切酶的作用下切割为核酸片段,核酸片段在连接酶的作用下得到酶切连接产物。其中,纯化采用现有技术已知的方法。
23.将酶切连接产物插入质粒载体之前需要对酶切连接产物进行纯化。其中,纯化采用现有技术已知的方法。
24.所述质粒载体与所述sgrna的比例是指数量的比值,即质粒载体的数量与sgrna的数量的比值。在一个具体的实施方式中,所述质粒载体与所述sgrna的比例为1-10:1、2-9:1、3-8:1、4-7:1或5-6:1,例如可以为1:1、2:1、3:1、4:1、5:1、6:1、7:1、8:1、9:1、10:1,优选为4:1、5:1、6:1、7:1或8:1。
25.在本文中使用的质粒载体没有具体的限定,只要是可以用于电击转化的载体即可,例如可以为腺病毒载体、腺相关病毒载体、逆转录病毒载体以及满病毒载体等;对于载体的大小也没有明确的限定,例如较小的腺相关病毒载体,可容纳5 kb大小的基因片段;慢病毒载体可容纳9 kb大小的基因片段。
26.在一个具体地实施方式中,其中在步骤3)中,其中所述质粒载体的浓度为30 ng/
µ
l-180 ng/
µ
l、40 ng/
µ
l-170 ng/
µ
l、50 ng/
µ
l-160 ng/
µ
l、60 ng/
µ
l-160 ng/
µ
l、70 ng/
µ
l-160 ng/
µ
l、80 ng/
µ
l-150 ng/
µ
l、90 ng/
µ
l-150 ng/
µ
l、100 ng/
µ
l-140 ng/
µ
l,例如可以为30 ng/
µ
l、40 ng/
µ
l、50 ng/
µ
l、60 ng/
µ
l、70 ng/
µ
l、80 ng/
µ
l、90 ng/
µ
l、100 ng/
µ
l、110 ng/
µ
l、120 ng/
µ
l、130 ng/
µ
l、140 ng/
µ
l、150 ng/
µ
l、160 ng/
µ
l、170 ng/
µ
l、180 ng/
µ
l,优选为90 ng/
µ
l-150 ng/
µ
l。在步骤3)中,所述质粒载体的浓度具体指的是步骤3)中,所述质粒载体在反应体系中的浓度。
27.在一个具体的实施方式中,所述电击转化的电压为大于1.0kv且小于2.0kv,例如可以为1.1kv、1.2kv、1.3kv、1.4kv、1.5kv、1.6kv、1.7kv、1.8kv、1.9kv,优选为1.4kv-1.8kv。例如为1.4kv、1.5kv、1.6kv、1.7kv、1.8kv。
28.本发明还提供上述方法制备的sgrnas质粒文库。
29.本发明还提供包含上述方法制备的sgrnas质粒文库的组合物。进一步地,该组合物为粒文库的储存液。
30.本发明提供的高效构建sgrnas质粒文库的方法可以显著降低文库构建时的工作量,尤其是当酶切连接反应体系中质粒载体与sgrna的比例为4:1到8:1时,纯化浓缩产物浓度为90ng/
µ
l到150ng/
µ
l时,电击转化反应时电压为1.4kv-1.8kv时,所得到的细菌克隆数能够有效提升质粒文库构建的覆盖度,为sgrna质粒文库构建提供了稳定高效的方法。
31.实施例
32.1. 酶切连接反应1.1 sgrna序列合成:sgrna序列采用外包服务的方式,通过invitrogen引物合成平台进行合成,合成的sgrna序列,结构如图1所示,该合成序列包含扩增用引物、bsmbi酶切位点以及20bp sgrna序列,具体的该合成序列如seq id no:1所示为:ttgtggaaaggacgaaaccgtctcaaccggacctgtgctgacaccacaggttttgagacgtagagctagagacagca。
33.序列扩增:1.2.1 将上述合成序列用去离子水稀释至10um后,作为sgrna扩增模板,按照表1所示将5
×
phusion hf buffer(thermo,#m0530l)、2.5 mm dntp(全式金,#ad101-01)、10 μm正向引物(invitrogen)、10 μm反向引物(invitrogen)(表2)、sgrna扩增模板(invitrogen)、phusion dna polymerase(thermo,#m0530l)以及去离子水(invitrogen,#10977015)进行混合加样。其中,扩增加样体系中,正向引物序列如seq id no:2所示,反向引物序列如seq id no:3所示。
34.表1 sgrna序列扩增加样体系表2 sgrna序列扩增用引物信息1.2.2 将上述sgrna扩增体系置于pcr扩增仪中,设定pcr反应程序为:98℃ 30s,1个循环;98℃ 10s,60℃ 20s,72℃ 15s,20个循环;72℃ 10min,1个循环;4℃保存,进行pcr扩增反应。
35.序列扩增产物纯化:运用pcr产物纯化试剂盒(天根,#dp214)根据该试剂盒说明书的描述,对sgrna pcr产物进行纯化。
36.1.3.1 将吸附柱放入收集管中,向吸附柱中加入500
µ
l平衡液,12000rpm离心1min后,倒掉收集管中的废液,将吸附柱重新放回收集管中。
37.1.3.2 取200
µ
l sgrna序列扩增产物,向其中加入3倍体积600
µ
l结合缓冲液,充分混匀后,将溶液加入吸附柱中,室温放置2min,12000rpm离心1min,倒掉收集管中的废液,将吸附柱放入收集管中。
38.1.3.3 向吸附柱中加入600
µ
l漂洗液(使用前检查是否已加入无水乙醇),
12000rpm离心1min,倒掉收集管中的废液,将吸附柱放入收集管中。重复该步骤一次。
39.1.3.4 将吸附柱放回收集管中,12000rpm离心2min,尽量除去漂洗液。将吸附柱置于室温放置数分钟,彻底晾干。与此同时,将去离子水在70℃提前进行预热,1.3.5将吸附柱放入一个干净的离心管中向吸附膜中间位置悬空滴加30
µ
l预热的去离子水,室温放置2min,12000rpm离心2min,收集pcr纯化产物。
40.1.3.6 取1.5
µ
l pcr纯化产物进行nanodrop 2000(thermofisher)检测,得到pcr纯化产物的浓度。
41.酶切连接反应:1.4.1 用去离子水将sgrna pcr纯化产物稀释至10ng/
µ
l,将质粒载体(北京大学魏文胜实验室赠与,载体的制备参考zhu, s., cao, z., liu, z. et al. guide rnas with embedded barcodes boost crispr-pooled screens. genome biol 20, 20 (2019))稀释至100ng/
µ
l后,按照表3所示将bsmbi (thermo, #er0452)、t4 dna ligase(neb, #m0202l)、dtt (索莱宝, #d1070)、atp(sigma, #a1825)、tango buffer(thermo, #er0452)以及质粒载体和sgrna进行混合加样。
42.表3 酶切连接反应加样体系1.4.2 将上述酶切连接体系置于pcr扩增仪中,设定反应程序为:37℃ 5min,16℃ 5min,20个循环;37℃ 5min,1个循环;4℃保存,进行酶切连接反应。1.5 酶切连接产物纯化:运用dna浓缩试剂盒(zymo research,#d4013)对酶切连接产物进行纯化浓缩。
43.1.5.1 取250
µ
l酶切连接产物加入1.5ml离心管中,同时加入5倍用量的dna结合缓冲液,迅速用枪头吹吸混匀。
44.1.5.2 将上述混合液加入zymo-spin
tm column中,并置于收集管中,室温12000rpm离心30s后,弃废液。
45.1.5.3 在zymo-spin
tm column中加入200
µ
l dna清洗液,室温12000rpm离心30s后,弃废液。
46.1.5.4 再次加入200
µ
l dna清洗液进行二次清洗后,室温12000rpm离心30s后,弃
废液。
47.1.5.5 将zymo-spin
tm column置于一个新的离心管中,室温放置几分钟,彻底晾干。与此同时,将去离子水在70℃提前进行预热。
48.1.5.6 在吸附膜中间位置悬空滴加10
µ
l预热的去离子水,室温放置2min后,12000rpm离心1min,收集纯化浓缩的酶切连接产物。
49.1.5.7 取1.5
µ
l pcr纯化产物进行nanodrop 2000(thermofisher)检测,得到酶切连接纯化浓缩产物的浓度。
50.1.5.8 将上述纯化浓缩产物分别稀释成30 ng/
µ
l、60 ng/
µ
l、90 ng/
µ
l、120 ng/
µ
l、150 ng/
µ
l,用于后续的电击转化实验。
51.电击转化:1.6.1 取1
µ
l纯化浓缩的酶切连接产物置于1.5ml离心管中,加入50
µ
l电击转化用大肠杆菌感受态hst08(takara,#9028)轻轻混匀后,用移液枪将预混液注入1mm孔径的电击杯(bio-rad,#1652093)中。
52.1.6.2 按照表4所示,设置电击转化条件(bio-rad,#gene p
µ
lser xcell)。表4 电击转化设定条件1.6.3 将电击杯置于电转仪卡槽中,进行电击转化。
53.1.6.4 电击转化反应结束后,在电击杯中迅速加入950
µ
l未添加抗生素的soc培养基,用移液枪吹打混匀后,转移到14 ml圆底离心管(falcon,# 352059)中,置于37℃,225rpm摇床中培养1h。其中,soc培养基包含1l sob、5 ml mgcl
2 (2m)、20 ml 葡萄糖 (1m)。
54.1.6.5 取1
µ
l菌液置于1.5ml离心管中,加入30
µ
l soc培养基轻轻混匀后,涂于带有抗性的15cm lb细菌培养板上,置于37℃培养箱中培养过夜。
55.1.6.6 统计培养板上的细菌克隆数,评估电击转化效率。
56.质粒文库构建1.7.1 运用1.1到1.6所描述的方法进行泛素化酶相关的质粒文库构建其中,根据uucd(http://uucd.biocuckoo.org)数据库提供的信息,针对422个泛素化酶编码基因共设计了5356条sgrnas,将设计好的sgrna插入到图1显示的sgrna的位置,从而构建了具有5356条sgrna的泛素化酶相关基因的质粒文库。
57.1.7.2 将1.6.5操作后剩余的菌液转移到lb培养基中,置于37℃,225rpm摇床中培养过夜后,8000 rpm离心5min,去上清,收集菌体,运用无内毒素质粒大提试剂盒(天根,#dp117)。
58.1.7.2 向留有菌体沉淀的离心管中加入含有rna酶a的细菌重悬液中,使用涡旋振荡器彻底悬浮细菌细胞沉淀。
59.1.7.3 向离心管中加入等体积的细菌裂解液,立刻上下温和的反转6-8次,使菌体
充分裂解,室温放置5min。
60.1.7.4 向离心管中加入8ml中和液,立即温和的上下翻转6-8次,充分混匀,至溶液出现白色分散絮状沉淀,然后室温放置10min左右,8000rpm离心10min,使白色沉淀沉积于管底,将全部溶液小心的倒入过滤器中,收集滤液。
61.1.7.5 向滤液中加入0.3倍滤液体积的异丙醇,上下颠倒混匀后转移至吸附柱中。室温8000rpm离心2min,倒掉收集管中的废液,将吸附柱重新放回收集管中。
62.1.7.6 向吸附柱中加入10ml漂洗液后,8000rpm离心2min,弃掉收集管中的废液,将吸附柱重新放回收集管中,重复操作该步骤一次。
63.1.7.7 向吸附柱中加入3ml无水乙醇,室温8000rpm离心2min,倒掉弃液。
64.1.7.8 将吸附柱重新放回收集管中,8000rpm离心5min,将吸附柱中残余的漂洗液去除。
65.1.7.9 将吸附柱置于一个干净的收集管中,向吸附膜中间位置滴加1-2ml洗脱缓冲液,室温放置5min后,8000rpm离心2min。
66.质粒文库质量检测1.8.1 取1-2
µ
l质粒提取物作为模板,按照表5所示将其与q5高保真酶混合液(neb; #m0492l)、10μm正向引物(seq id no:4:tatcttgtggaaaggacgaaacacc ,invitrogen)、10μm反向引物(seq id no:5:aatacggttatccacgcggc,invitrogen)和去离子水(invitrogen,#10977015)混合。将该反应体系置于pcr扩增仪中,设定pcr反应程序为:98℃ 30s,1个循环;98℃ 10s,60℃ 20s,72℃ 15s,20个循环;72℃ 10min,1个循环;4℃保存,进行pcr扩增反应。
67.表5 pcr扩增加样体系1.8.2 针对稳定插入的sgrna序列部分进行pcr扩增反应后,将pcr产物进行二代测序,通过生物信息学方法对文库中sgrna的丢失情况以及分布情况进行分析,评估该方法在细胞文库构建中的可行性和可靠性。
68.研究结果:对于sgrna质粒文库的构建而言,保障sgrna质粒文库的覆盖度(即在sgrna表达载体进行电击转化时,能够代表单个sgrna的平均菌落数)是确保sgrna质粒文库质量的关键。在整个工艺流程中,能够最终影响文库覆盖度的关键因素包括酶切连接反应体系中质粒与sgrna的比例、纯化浓缩产物浓度以及电击转化条件等。在这三个因素中,酶切连接反应中质粒与sgrna的比例对文库构建的影响会通过纯化回收产物浓度以及该产物在不同电转条件下所得的菌落克隆数得以直观体现。因此,该工艺流程的优化针对上述三个关键因素及其组合进行研究,最终寻找到最优化的方案。
69.酶切连接比例对电击转化效率的影响在该反应体系中所使用的质粒骨架中有细菌致死基因,也就是说在酶切连接反应中,只有当sgrna片段成功替代了细菌致死基因之后,才能在电击转化反应后形成可生长的细菌克隆。因此,酶切连接比例通过影响酶切连接反应的效率,进而影响电击转化反应后所形成的克隆数。作为对照组,当质粒骨架与sgrna的比例为1:0时,不能进行实质性的酶切连接反应,所以,在电转反应之后不能观察到大量克隆数的形成。该现象证明整体工艺流程下的结果真实可靠。基于此,当质粒骨架与sgrna的比例从1:1和2:1逐渐上升到4:1和6:1时,电击转化反应后所形成的克隆数呈现显著的上升趋势。并且当质粒骨架与sgrna的比例为1:1和2:1时,电击转化所形成的克隆数显著较低,证明该反应体系下的酶切反应效率普遍偏低。但是质粒骨架与sgrna的比例上升到8:1之后,电击转化反应后所形成的克隆数基本趋于平缓,不再有显著性的上升态势。因此,电击转化反应后所形成的克隆数随着酶切连接反应中质粒骨架与sgrna比例的增加呈现整体上升的趋势。但是,当酶切反应体系中质粒骨架与sgrna比例达到一定的峰值后,电击转化后所形成的克隆数将维持不变(图2-图4)。因此,当酶切连接比例为4:1到8:1时,电击转化后产生的克隆数可以有效提升质粒文库构建的效率。
70.纯化浓缩产物浓度对于电击转化效率的影响纯化浓缩产物的浓度会直接影响电击转化反应后所形成的克隆数。作为对照组,质粒骨架与sgrna的比例为1:0,无论纯化浓缩产物浓度高低,因为并不能发生实质性的酶切连接反应,所以电击转化反应后未能观察到大量的克隆数。当质粒骨架与sgrna的比例为1:1和2:1时,随着纯化浓缩产物浓度的上升,在电击转化反应后所形成的克隆数也呈现明显的上升趋势。但整体而言,电击转化克隆数仍未达到最佳状态,有很大的提升空间。当质粒骨架与sgrna的比例为4:1、6:1以及8:1时,随着纯化浓缩产物浓度的上升,电击转化反应后所形成的克隆数上升到一定的峰值之后,进入平台期(图2-图4)。相比之下,当质粒浓度达到90 ng/
µ
l到150 ng/
µ
l时,电击转化所得到的克隆数可以有效提升质粒文库构建的效率。
71.电击转化条件对于电击转化效率的影响电击转化是通过在细菌细胞膜上瞬间成孔,使得外源质粒进入细菌细胞内,完成转化过程的方法。因此,电击转化的条件对于提升转化效率至关重要,而电击转化条件的调整也基本上体现在由电压造成的电场场强的改变上。在该实验中,申请人测试了1.0kv、1.4kv、1.8kv以及2.0kv四个电转条件。结果显示,当电压为1.0kv时,电击转化反应后所形成的克隆数普遍偏低,表明该条件并不能进行高效的电击转化反应。但是当电压为2.0kv时,在实验过程中会观察到放电现象,最终所得的数据无法进行有效的统计,表明该条件不适合进行稳定有效的电击转化反应。当电压为1.4kv以及1.8kv时,可进行高效的电击转化反应,并得到较高的克隆数,在其他条件较优化的状态下,克隆数可稳定维持在2500-3500之间,表明该方法在有效提升质粒文库构建覆盖度的同时,大量降低质粒文库构建时的工作量(图2-图4)。
72.酶切连接比例、纯化浓缩浓度以及电击转化条件组合对于电击转化效率的影响电击转化效率受酶切连接比例、纯化浓缩浓度以及电击转化条件等因素的综合影响。其中,酶切连接反应中质粒骨架与sgrna的比例影响酶切连接反应效率,进而影响在酶
切连接反应体系中形成的真正能够表达sgrna载体的比例;而纯化浓缩浓度关系到一定体积内所包含的质粒数量;电击转化条件通过影响质粒进入细菌内的效率进而影响最终观察到的细菌克隆数。在对照反应中,因为酶切反应体系中只有质粒骨架,并未加入sgrna片段,因此,质粒骨架即便被成功酶切,也并不能形成完整的表达质粒;与此同时,未能酶切的质粒因为其中插入细菌自杀基因,在电击转化反应后也并不能形成完好的克隆。因此,该对照组所表现出来的极个别的细菌克隆数(允许一定的误差)为正常现象,证明该体系所呈现的结果真实可靠。在此基础上,当质粒骨架与sgrna的比例为1:1和2:1时,随着纯化浓缩产物浓度的增加,在电击转化反应后所得到的克隆数也呈现整体上升趋势,但是克隆数仍未达到最佳优化效果。当质粒骨架与sgrna的比例为4:1、6:1以及8:1时,随着纯化浓缩产物浓度的增加,在电压为1.4kv-1.8kv时,所得到的克隆数可达2500-3500个,能够显著提升质粒文库的构建效率(图2-图4)。
73.5. 质粒文库质量评估运用质粒文库构建的优化方法构建泛素化酶相关的质粒文库,运用二代测序的方法,通过检测sgrna的丢失个数、基因丢失个数以及sgrna分布情况评估质粒文库的质量。结果显示,该文库中的5356个sgrna没有出现sgrna丢失和基因丢失(表6),且sgrna呈现均一的正态分布情况(图5)。
74.表6 泛素化酶相关质粒文库质量评估参数泛素化酶相关基因文库评估参数平均测序深度473xsgrna丢失个数0基因丢失个数0sgrna总数5356基因总数422结论:在该发明中,sgrna质粒文库的构建需进行酶切连接反应、纯化浓缩过程以及电击转化反应三个过程,在各个反应过程中,酶切连接反应体系中质粒载体与sgrna的比例、纯化浓缩产物浓度以及电击转化反应过程中的电转条件均能对质粒文库构建的质量和效率产生影响,最直观的参数即为电击转化反应后所得到的菌落克隆数。在优化质粒文库的构建方法时发现,当酶切连接反应体系中质粒载体与sgrna的比例为4:1到8:1时,纯化浓缩产物浓度为90ng/
µ
l到150ng/
µ
l时,电击转化反应时电压为1.4kv-1.8kv时,所得到的细菌克隆数能够有效提升质粒文库构建的覆盖度,同时显著降低文库构建时的工作量,为sgrna质粒文库构建提供了稳定高效的方法。