1.本发明属于有机发光材料领域,主要涉及一类含氮杂芳环衍生物以及羰基构筑的热激活延迟荧光材料,其具有窄半峰宽发射,发射波长可随给电子基团强度可调的特征,将其作为客体运用于有机电致发光器件,可得高性能的有机电致发光二极管。
背景技术:
2.热激活延迟荧光材料理论上能够实现100%的内量子效率,打破了传统荧光器件外量子效率5%的理论极限,并且可以与贵金属磷光配合物器件相媲美,被认为是继荧光材料和磷光材料之后的第三代有机电致发光二极管材料。但是传统的热激活延迟荧光(tadf)分子是通过减小homo与lumo的重叠,进而减小最低单线态(s1)与最低三线态(t1)间的能级差,从而实现t1到s1间的反系间窜越(risc),达到充分利用三线态激子的目的。这也导致分子的振子强度较小,分子的发光量子效率相对较低,较大的半峰宽。直到多重共振热激活延迟荧光(mr-tadf)材料的出现,由于其激发态结构弛豫被刚性骨架所抑制,从而获取极窄的半峰宽,同时短程的电荷转移表现出高的振子强度,因此进一步提高荧光量子产率。mr-tadf材料同时兼顾高发光效率以及高色纯度的特征使其成为有机电致发光材料的重要研究方向。
技术实现要素:
3.多重共振热激活延迟荧光材料自开发以来,已经在蓝光、绿光以及红光乃至深红光取得进展,然而在氮杂芳环衍生物中嵌入硼原子合成难度较大,且不易调控其光色。截至目前mr-tadf分子主要围绕于硼/氮体系,而对于其他骨架的研究很少,因此开发更多骨架、研究其他骨架材料的发光特性以及其在有机发光二极管中的应用具有重要意义。
4.本发明旨在开发氮杂芳环衍生物与羰基构筑的tadf材料作为光色易调、高色纯度、高激子利用率的有机发光材料以及其在有机电致发光二极管中的应用。这类材料合成方法简单,刚性骨架与短程电荷转移可以同时提高色纯度和发光效率。因此该类氮杂芳环衍生物与羰基构筑的tadf材料将是一类具有竞争力的新型有机发光材料。
5.一种含氮杂芳环衍生物与羰基构筑的tadf材料,其化学结构如式1所示,
[0006][0007]
r1、r2和r3分别独立为氢、芳基、杂芳基、二芳基氨基、芳基杂芳基氨基、烷基、烷氧
基或芳氧基。
[0008]
为
[0009]
含氮杂芳环衍生物与羰基构筑的tadf材料,优选的化学结构如下式所示
[0010][0011]
本发明的另外一个目的是提供氮杂芳环衍生物与羰基构筑的tadf材料的应用,氮/羰基类tadf材料用于发光领域,将其作为有机电致发光二极管的客体发光材料,用于制备真空蒸镀型有机电致发光二极管。
[0012]
本发明的有益效果为:
[0013]
与现有技术相比,本发明的有益效果在于:本发明的氮杂芳环衍生物与羰基构筑的tadf材料具有合成简单、稳定的结构、高发光效率、较小的半峰宽等优势。本发明不同于硼/氮体系的mr-tadf分子,该材料的合成避开使用危险的有机锂试剂,涉及的乌尔曼反应、水解反应、傅克酰基化反应安全简单;具有硼/氮体系的mr-tadf分子同样的优势窄半峰宽与高色纯度;改变氮杂芳环衍生物,调整其给电子能力,可以轻易调节发光谱图位置。
附图说明:
[0014]
【图1】为本发明实施例1、例2制得的化合物2、5、8、11分别在甲苯溶液中的紫外可见光吸收光谱图。
[0015]
【图2】为本发明实施例1、例2制得的化合物2、5、8、11分别在甲苯溶液中的光致发光光谱图。
[0016]
【图3】为本发明实施例1、例2制得的化合物2、5、8、11分别在不同溶液中的光致发光光谱图。
[0017]
【图4】为本发明实施例1、例2制得的化合物2、5、8、11的电致发光光谱。
[0018]
【图5】为本发明实施例1、例2制得的化合物2、5、8、11的器件性能表征图。
具体实施方式
[0019]
为了更清楚的说明本发明,下面结合优选实施例和附图对本发明作进一步说明。本领域技术人员应当理解,下面所描述的具体内容是说明性的而非限制性的,不能因此限制本发明的保护范围。
[0020]
本发明中,制备方法如无特殊说明均为常规方法。所用的原料如无特别说明均可以从公开的商业途径获得。
[0021]
实施例1
[0022]
本发明提供的式(1)中,当r1为叔丁基,r2和r3为h,为时,为下式化合物2,其合成路线如下:
[0023][0024]
化合物1的合成:
[0025]
将叔丁基咔唑(13.43g,48mmol)、叔丁基碘苯(15.00g,58mmol)、碘化亚铜(0.91g,4.8mmol)、邻菲罗玲(1.73g,9.6mmol)、碳酸钾(19.87g,14.4mmol)、18-冠醚-6(1.90g,7.2mmol)加入到二氧六环(60ml)中,氮气保护110℃下反应过夜。冷却,旋干,用二氯甲烷/水萃取,有机相用无水硫酸钠干燥,浓缩后硅胶柱层析(石油醚:二氯甲烷=5:1)得到白色固体17.91g,收率90.68%。1h nmr(500mhz,cdcl3)δ8.14(d,j=1.7hz,2h),7.57(d,j=8.5hz,2h),7.48
–
7.42(m,4h),7.35(d,j=8.6hz,2h),1.46(s,18h),1.41(s,9h).
13
c nmr(101mhz,cdcl3)δ149.91,142.61,139.39,135.47,126.63,126.25,126.22,123.52,123.26,116.21,109.38,34.79,32.13,31.53,31.50.
[0026]
化合物2的合成:
[0027]
将化合物1(4.93g,12mmol),1,1-二氯二甲基醚(1.93g,16.8mmol)加入到邻二氯苯(20ml)中。氮气保护0℃下加入ticl4(3.64g,19.2mmol),移至室温反应4h。向体系内加入hcl淬灭反应,用二氯甲烷/水萃取,有机相用无水硫酸钠干燥,浓缩后硅胶柱层析(石油醚:二氯甲烷=2:1)得到淡黄色固体1.84g,收率35%。1h nmr(400mhz,cdcl3)δ8.69(d,j=2.3hz,1h),8.51(d,j=1.2hz,1h),8.45(d,j=1.5hz,1h),8.36(d,j=8.9hz,1h),8.24
–
8.15(m,2h),7.92(dd,j=8.8,2.3hz,1h),7.66(dd,j=8.7,1.7hz,1h),1.55(s,9h),1.50(s,9h),1.47(s,9h).
13
c nmr(101mhz,cdcl3)δ179.11,146.48,146.10,146.01,137.65,137.52,137.29,131.60,126.52,125.24,125.13,125.01,124.74,122.55,121.02,119.08,117.97,115.26,113.38,35.46,34.88,34.79,32.05,31.79,31.43.
[0028]
实施例2
[0029]
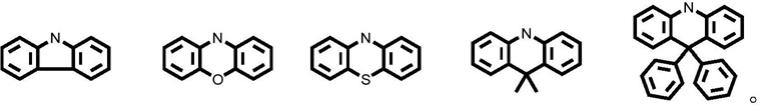
本发明提供的式(1)中,当r1,r2和r3为h,分别为分别为时,为下式化合物5,8,11,其合成路线如下:
[0030][0031]
化合物3的合成:
[0032]
将吖啶(4.18g,20mmol)、2-碘苯甲酸甲酯(5.76g,22mmol)、碘化亚铜(381mg,2mmol)、铜(1.3mg,20mmol)和碳酸钾(3.03g,22mmol)加入到二氯苯(30ml)中,氮气保护下190℃下反应24h。冷却,过滤,二氯甲烷洗涤固体,滤液浓缩后硅胶柱层析(石油醚:二氯甲烷=2:1)得到浅绿色固体5.89g,收率85.8%。1h nmr(400mhz,cdcl3)δ8.20
–
8.14(m,1h),7.76(td,j=7.7,1.5hz,1h),7.63
–
7.56(m,1h),7.45(dd,j=7.5,1.5hz,2h),7.34(d,j=7.8hz,1h),6.97
–
6.85(m,4h),6.05(d,j=9.1hz,2h),3.54(s,3h),1.71(d,j=22.8hz,6h).
13
c nmr(126mhz,cdcl3)δ165.95,140.70,140.43,134.58,133.41,132.74,132.35,129.59,128.56,126.37,125.64,120.35,113.53,52.34,35.87,33.24,31.81.
[0033]
化合物4的合成:
[0034]
将化合物3(5.83g,17mmol)和氢氧化锂(1.22g,51mmol)溶于四氢呋喃/甲醇/水(40ml,v/v/v=3:1:1)中,70℃下搅拌16h,浓缩部分溶剂,1m的盐酸调ph到3,析出淡黄色固体,抽滤,干燥,得到5.5g,收率98.3%。
[0035]1h nmr(400mhz,cdcl3)δ8.28(dd,j=7.9,1.3hz,1h),7.79(td,j=7.7,1.5hz,1h),7.64
–
7.56(m,1h),7.44(dd,j=5.9,3.4hz,2h),7.31(d,j=7.8hz,1h),6.91(dd,j=6.0,3.4hz,4h),6.02(d,j=9.4hz,2h),1.79(s,3h),1.50(s,3h).
13
c nmr(126mhz,cdcl3)δ142.08,140.16,135.62,133.53,133.24,130.35,128.81,126.53,125.93,121.15,114.22,35.81,34.35,31.21.
[0036]
化合物5的合成:
[0037]
将化合物4(4.9g,15mmol)溶于二氯甲烷(30ml),向溶液中滴加2d dmf后,0℃下慢慢滴加草酰氯(9.52g,75mol),滴毕,转至室温下反应2h至其全部溶解,减压除去溶剂,再次加入30ml二氯甲烷,然后向体系中加入三氯化铝(8g,60mmol)室温反应30min。将体系慢慢滴进50ml稀盐酸(1m),用二氯甲烷萃取,有机相用无水硫酸钠干燥,浓缩后硅胶柱层析(石油醚:二氯甲烷=1:1),得到黄色固体3.1g,收率66.4%。1h nmr(400mhz,cdcl3)δ8.53
–
8.48(m,1h),8.34
–
8.28(m,1h),8.04(d,j=8.5hz,1h),7.78
–
7.73(m,1h),7.69
–
7.57(m,3h),7.39(td,j=7.8,2.8hz,2h),7.23(dd,j=7.5,5.6hz,2h),2.01(s,3h),1.36(s,3h)
.
13
c nmr(101mhz,cdcl3)δ179.30,140.16,139.16,137.43,137.24,135.27,132.31,127.59,127.46,126.29,125.41,125.32,124.44,124.42,123.39,123.27,123.13,119.82,119.44,37.03,31.08,23.05.
[0038]
化合物6的合成:
[0039]
将吩噁嗪(5.49g,30mmol)、2-碘苯甲酸甲酯(8.65g,33mmol)、碘化亚铜(0.57mg,3mmol)、铜(1.95mg,30mmol)和碳酸钾(3.56g,33mmol)加入到二氯苯(30ml)中,氮气保护下190℃下反应24h。冷却,过滤,二氯甲烷洗涤固体,滤液浓缩后硅胶柱层析(石油醚:二氯甲烷=2:1)得到黄色固体7.29g,收率76.5%。1h nmr(400mhz,cdcl3)δ8.18
–
8.10(m,1h),7.78
–
7.71(m,1h),7.56(t,j=7.6hz,1h),7.40(d,j=7.7hz,1h),6.69
–
6.52(m,6h),5.78(d,j=9.0hz,2h),3.71(s,3h).
13
c nmr(126mhz,cdcl3)δ165.78,143.86,138.53,134.69,134.07,133.33,133.12,131.65,128.87,123.18,121.26,115.41,112.90,52.55.
13
c nmr(126mhz,cdcl3)δ167.67,144.08,140.08,135.79,133.65,132.84,130.37,129.07,123.32,122.02,115.64,113.87.
[0040]
化合物7的合成:
[0041]
将化合物6(6.36g,20mmol)和氢氧化锂(1.43g,60mmol)溶于四氢呋喃/甲醇/水(60ml,v/v/v=3:1:1)中,70℃下搅拌16h,浓缩部分溶剂,1m的盐酸调ph到3,析出黄色固体,抽滤,干燥,得到6g,收率99%。1h nmr(400mhz,cdcl3)δ8.29(d,j=7.8hz,1h),7.84
–
7.74(m,1h),7.58(t,j=7.6hz,1h),7.42(d,j=7.8hz,1h),6.78
–
6.62(m,4h),6.62
–
6.48(m,2h),5.81(d,j=7.8hz,2h).
13
c nmr(126mhz,cdcl3)δ167.67,144.08,140.08,135.79,133.65,132.84,130.37,129.07,123.32,122.02,115.64,113.87.
[0042]
化合物8的合成:
[0043]
将化合物7(1.5g,5mmol)溶于二氯甲烷(30ml),向溶液中滴加2d dmf后,0℃下慢慢滴加草酰氯(3.17g,25mol),滴毕,转至室温下反应2h至其全部溶解,减压除去溶剂,再次加入30ml二氯甲烷,然后向体系中加入三氯化铝(2.67g,20mmol)室温反应30min。将体系慢慢滴进50ml稀盐酸(1m),用二氯甲烷萃取,有机相用无水硫酸钠干燥,浓缩后硅胶柱层析(石油醚:二氯甲烷=1:1),得到黄色固体640mg,收率45%。1h nmr(400mhz,cdcl3)δ8.52
–
8.46(m,1h),8.05(d,j=8.6hz,1h),7.97(dd,j=6.6,2.8hz,1h),7.67(ddd,j=8.6,7.2,1.5hz,1h),7.58(d,j=8.1hz,1h),7.37(t,j=7.5hz,1h),7.24
–
7.17(m,2h),7.14(d,j=4.0hz,2h),7.07(dt,j=8.8,4.4hz,1h).
13
c nmr(101mhz,cdcl3)δ177.62,148.25,146.28,138.35,133.16,132.62,128.35,127.88,126.10,124.81,123.78,123.74,123.40,123.29,120.62,118.26,118.19,118.06,117.62.
[0044]
化合物9的合成:
[0045]
将吩噻嗪(1.75g,8.78mmol)、2-碘苯甲酸甲酯(2.3g,8.78mmol)、碘化亚铜(167mg,0.88mmol)和碳酸钾(1.21g,8.78mmol)加入到二氯苯(30ml)中,氮气保护下190℃下反应24h,冷却,过滤,二氯甲烷洗涤固体,滤液浓缩后硅胶柱层析(石油醚:二氯甲烷=2:1)得到白色固体2.44g,收率83.2%。1h nmr(400mhz,cdcl3)δ8.16(dd,j=7.8,1.2hz,1h),7.76(td,j=7.7,1.5hz,1h),7.58(dd,j=11.1,4.2hz,1h),7.46(d,j=7.8hz,1h),6.95(dd,j=6.8,2.2hz,2h),6.79-6.72(m,4h),5.97(dd,j=7.7,1.5hz,2h),3.73(s,3h).
13
c nmr(75mhz,cdcl3)δ166.12,143.52,140.00,134.29,133.77,132.91,132.24,128.84,
126.76,126.53,122.23,119.20,115.16,52.56.
[0046]
化合物10的合成:
[0047]
将化合物9(1.66g,5.0mmol)和氢氧化锂(0.36g,15.0mmol)溶于四氢呋喃/甲醇/水(40ml,v/v/v=3:1:1)中,室温下搅拌16h,浓缩部分溶剂,1m的盐酸调ph到5,析出淡黄色固体,抽滤,干燥,得到1.48g,收率93.1%。1h nmr(400mhz,cdcl3)δ8.40(d,j=7.8hz,1h),8.40(d,j=7.8hz,1h),7.83(t,j=7.0hz,1h),7.64(t,j=7.6hz,1h),7.53(d,j=7.8hz,1h),7.09
–
7.06(m,2h),6.87
–
6.84(m,4h),6.14
–
6.12(m,2h).
13
c nmr(75mhz,cdcl3)δ167.23,143.37,140.60,134.98,134.10,133.62,129.97,129.22,126.94,126.91,123.23,120.87,116.04
[0048]
化合物11的合成:
[0049]
将化合物10(1.17g,3.66mmol)溶于二氯甲烷(30ml),向溶液中滴加2d dmf后,慢慢滴加草酰氯(2.32g,18.25mol),滴毕,转至室温下反应2h,然后向反应体系中加入三氯化铝(1.95g,14.6 2mmol)转至油浴中回流反应12h,冷却,向体系中慢慢滴加1m稀盐酸10ml,二氯甲烷萃取,饱和氯化铵水溶液洗涤,有机相用无水硫酸钠干燥,浓缩,硅胶柱层析(石油醚:二氯甲烷=3:1),得到淡黄色固体0.72g,收率65.1%。1h nmr(400mhz,cdcl3)δ8.43(d,j=8.0hz,1h),8.20(d,j=7.9hz,1h),7.84(d,j=8.5hz,1h),7.63(t,j=7.8hz,1h),7.54(d,j=7.4hz,1h),7.41-7.38(m,2h),7.35
–
7.29(m,2h),7.21
–
7.17(m,2h).
13
c nmr(75mhz,cdcl3)δ178.89,142.87,141.45,139.49,132.56,130.64,129.05,127.49,127.26,127.15,126.20,125.79,124.83,124.37,124.31,124.08,123.97,121.65,120.54.元素分析,理论:c,75.73;h,3.68;n,4.65;s,10.64。实际:c,75.75;h,3.66;n,4.66。
[0050]
本发明所涉及的新型氮杂芳环衍生物与羰基构筑的tadf化合物均可按照以上实施例所示类似的方法合成出来。所有目标分子均分子刚性,减小了分子振动,抑制了环间转动,进而得到色纯度高的发射峰;所得目标产物均大共轭平面,利于电子离域,增强振子强度,从而提高发光效率;更换不同的氮杂芳环衍生物,调控其给电子能力可以改变其发光颜色。本发明还提供了所涉及的该类化合物的紫外-可见吸收光谱以及光致发光光谱,以及在电致发光器件中的相关图谱信息。
[0051]
实施例3
[0052]
将化合物2、5、8、11溶解在甲苯中配成10-5
m溶液,测试其溶液的紫外可见吸收光谱。由图1可知,这三类化合物在溶液中的紫外可见吸收光谱大致有两种吸收峰:短波长(325nm)处的吸收峰主要归属于分子的π-π*的跃迁吸收;长波长(400~410nm)的吸收峰归属于分子内给体单元到受体单元的电荷转移(ict)跃迁吸收峰。
[0053]
实施例4
[0054]
将化合物2、5、8、11溶解在甲苯中配成10-5
m溶液,测试其溶液的光致发光光谱。如图2所示,化合物2、5、8、11在光激发下化合物的最大发射峰分别为431nm、447nm、485nm、501nm,半峰宽分别为36nm、61nm、76nm、86nm,斯托克斯位移分别为895cm-1
、2320cm-1
、3595cm-1
、4430cm-1
。可以看出随着分子内ct作用的增强,发光光谱明显红移,半峰宽与斯托克斯位移也明显增大。
[0055]
实施例5
[0056]
将化合物2、5、8、11分别溶解在正己烷、甲苯、乙醚、二氯甲烷、乙酸乙酯、丙酮、乙
腈和甲醇中配成10-5
m溶液,测试其溶液的光致发光光谱。如图3所示,化合物2、5、8、11从低极性溶剂到高极性溶剂展现了明显的发射峰红移特征,其中czao从正己烷到甲醇的最大红移达到了53nm,而dqao为76nm,qpxo为99nm,qpo为117nm。从最大红移量可以看出czao、dqao、qpxo和qpo的分子内ct作用逐渐增强。
[0057]
实施例6
[0058]
实施例1、例2中的化合物2、5、8、11在有机电致发光器件中的电致发光光谱发光光谱。以化合物2、5、8、11作为器件发光层掺杂剂制备结构为ito(110nm)/pedot:pss(35nm)/mcp(30nm)/化合物2、5、8、11:mcpcn(3wt%,30nm)/tmpypb(50nm)/lif(0.5nm)/al(120nm)的有机电致发光二极管。其中,pedot:pss为空穴注入层,mcp是空穴传输层,mcpcn为发光层主体材料,tmpypb为电子传输层,lif/al为阴极。其中,化合物2、5、8、11都是3wt%掺杂,如图4所示,获得的电致发光光谱与光致发光光谱一致,体现出不断红移的特征。
[0059]
实施例7
[0060]
实施例1、例2中的化合物2、5、8、11在有机电致发光器件中的电致发光性能。如图5所示,czao、dqao、qpxo和qpo其对应器件获得最大外量子效率分别为为8.62%、10.28%、7.05%和15.29%,最大功率效率分别为4.28lm w-1
、9.60lm w-1
、14.11lm w-1
和25.96lm w-1
,最高电流效率分别为5.46cd a-1
、11.02cd a-1
、16.20cd a-1
和45.56cd a-1
,可见随着分子内ct作用的增强,对应的功率效率和电流效率也都逐渐增大。
[0061]
尽管结合了优选实施例对本发明进行了说明,但本发明并不局限于上述实施例,应当理解所附权利要求概括了本发明的范围。在本发明构思的指导下,本领域的技术人员应当意识到,对本发明的各实施例方案所进行的一定的改变,都将被本发明的权利要求书的精神和范围所覆盖。