1.本发明涉及药物化学合成技术领域,更具体地,涉及一种麦角甾醇及其衍生物的制备方法。
背景技术:
2.麦角甾醇是生产维生素d2的前体,也是生产激素类药物的中间体,在紫外线照射下可被转化为维生素d2。麦角甾醇是从真菌类酵母与麦角菌中发现的一种植物固醇,是微生物细胞膜的重要组成部分,对确保细胞膜的完整性、膜结合酶的火星、膜的流动性、细胞活力以及细胞物质运输等起着重要作用。有研究显示,麦角甾醇可能具有抗肿瘤特性。
3.目前生产麦角甾醇的方法是从培养的酵母内提取,通过培养酵母菌体,破碎后提取麦角甾醇,收率非常低,废水量大,而且经该方法提取的麦角甾醇含有其他同系物,提取分离相对困难。
4.目前未有成熟合成的相关路线报道。
技术实现要素:
5.基于现有技术中存在的上述技术问题,本发明提供了一种麦角甾醇及其衍生物的制备方法,通过该方法可针对性获得麦角甾醇及其衍生物的目标结构,且收率高、纯度高。
6.为了实现上述目的,本发明的技术方案如下:
7.步骤1、wittig试剂的制备:
8.步骤1-1:化合物a0在盐酸二甲胺和甲醇水条件下,发生曼尼希反应得到化合物a1;
9.步骤1-2:所述化合物a1在甲醇溶剂下经催化氢化,再经硼氢化钠还原得到化合物a3;反应过程中,加入催化剂和阻聚剂,所述催化剂为雷尼镍,所述阻聚剂为2,6-二叔丁基对甲苯酚;氢化温度为30~60℃;
10.步骤1-3:所述化合物a3经过卤代反应得到化合物a4;
11.步骤1-4:所述化合物a4与磷试剂反应得到所述wittig试剂;
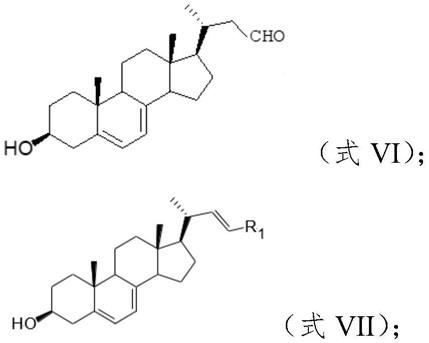
12.步骤2、将化合物vi与所述wittig试剂反应,得到麦角甾醇及其衍生物;
13.其中,所述化合物vi具有下式vi结构,所述麦角甾醇及其衍生物具有下式vii结构:
[0014][0015]
所述r1为中的一种;
[0016]
所述化合物a0的结构为:r
0-cho;
[0017]
其中,所述r0为为中的一种。
[0018]
具体地,步骤1中,以化合物a0为异戊醛为例,wittig试剂的制备路线如下:
[0019][0020]
在化合物a1制备a3过程中,一般会生成化合物a2和化合物a3,经后处理得到化合物a3。
[0021]
在一些实施方式中,所述磷试剂包括三苯基膦,亚磷酸三甲酯,亚磷酸三乙酯等中的至少一种;优选使用三苯基磷。
[0022]
在一些实施方式中,步骤1-4中,溶剂为甲苯,反应体系中还加入催化剂,所述催化剂质量为卤代异戊烷质量的1~5%,所述催化剂为碘化钾。在一些实施方式中,步骤1-2的反应过程中,加入的催化剂和阻聚剂,所述催化剂为雷尼镍,所述阻聚剂为2,6-二叔丁基对甲苯酚;氢化温度为30~60℃。在甲醇溶剂下升温并加阻聚剂和雷尼镍催化氢化;升温并以雷尼镍作为催化剂,可加快反应速率,减少聚合程度,提高化合物a3的收率;而加上阻聚剂,可进一步减少聚合成都,进而提高收率;氢化温度为30~60℃;优选氢化温度为55℃。
[0023]
在一些实施方式中,步骤1-4中,溶剂为甲苯,还加入催化剂,所述催化剂质量为卤代异戊烷质量的1~5%,所述催化剂为碘化钾。经发明人试验,不同的溶剂和水分对转化率有一定的影响,其中,甲苯分水的收率最高,而加入碘化钾作为催化剂,可提高转化率,并抑制异构体的产生,提高化合物wittig试剂的收率。
[0024]
在一些实施方式中,以化合物v为原料,经侧链醛基化,得到化合物vi;其中,所述化合物v具有以下式v结构:
[0025][0026]
其中,r0为离去基团。
[0027]
在一些实施方式中,所述化合物v在碱性条件下,经dmso高温氧化得到化合物vi。
[0028]
在一些实施方式中,所用碱为碳酸氢钠、吡啶、三乙胺、3-甲基吡啶、dmap、氢氧化钠、碳酸钠中的至少一种;反应温度为80~120℃。
[0029]
在一些实施方式中,所述化合物v由化合物i经侧链磺酰化、脱氢、酯化、还原脱氢制备而成;所述化合物i的结构式如下式i所示:
[0030][0031]
在一些实施方式中,化合物v的制备包括以下步骤:
[0032]
s1、在碱性条件下,在所述化合物i中加入甲苯黄酰氯进行反应,得到化合物ii;
[0033]
s2、所述化合物ii经脱氢反应得到化合物iii,脱氢过程为先醚化,然后加入脱氢试剂脱氢;
[0034]
s3、所述化合物iii在催化剂作用下与酯化试剂反应得到化合物iv;
[0035]
s4、所述化合物iv在碱性条件下经过硼氢化合物还原得到所述化合物v。
[0036]
在一些实施方式中,碱性条件为加入dmap、三乙胺、吡啶、三甲基吡啶中的至少一种;反应溶剂为二氯甲烷、氯仿、甲苯、乙酸乙酯、二氯乙烷中的至少一种;反应温度为-10~50℃。
[0037]
在一些实施方式中,步骤s2中,所述脱氢试剂为ddq和/或四氯苯醌。
[0038]
在一些实施方式中,步骤s3中,所述酯化试剂为酸酐、乙酰氯、醋酸异丙酯中的至少一种,所述催化剂为pts,hcl,h2so4,hclo4,msoh中的至少一种;和/或,反应温度为0~85℃。
[0039]
在一些实施方式中,步骤s4中,碱性条件为加入naoh,koh,naome,t-buok,吡啶,三乙胺,dmap中的至少一种;和/或,所述硼氢化合物包括ca(bh4)2、nabh4、kbh4中的至少一种。
[0040]
相较于现有技术,本发明的有益效果如下:
[0041]
本发明提供了一种麦角甾醇的制备方法,包括wittig试剂的制备、甾体骨架结构
的设计,最后将二者进行合成反应得到麦角甾醇或其衍生物。本发明创造性地以合成的方法,可针对性地获得特定结构的麦角甾醇或其衍生物,收率高(重量收率达90%)、纯度高(>99%),远高于现有技术的方法。另外,本发明的方法中,以醛类试剂作为原料,物料来源广泛,而且合成路线短,绿色环保,适于工业化生产。
[0042]
另外,通过本发明在制备过程中对工艺条件的优化,可极大地提高中间体的收率和纯度,从而提高麦角甾醇及其衍生物的纯度和收率,原料转化率高。
具体实施方式
[0043]
在下面的描述中阐述了很多具体细节以便于充分理解本发明。但是本发明能够以很多不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似改进,因此本发明不受下面公开的具体实施的限制。
[0044]
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。
[0045]
一、wittig试剂的制备
[0046]
以异戊醛(化合物a0)为原料,具体路线如下所示:
[0047][0048]
上述制备路线中,具体包括以下步骤:
[0049]
①
化合物a1的制备
[0050]
反应瓶中,加入0.8kg化合物a0,1.144kg盐酸二甲胺,1.1kg甲醛水溶液(40%),机械搅拌均匀,加热升温至70℃,搅拌回流反应4-7h。gc监测产物和原料比值,开始后处理。搭置水蒸气蒸馏装置,水蒸汽发生装置温度约130-150度,保证水蒸气气泡充分,接硬管对空;水蒸气通到反应液中,反应液温度约110度。适时补充水。馏出物为产品和水混合物,蒸馏约8h,至溜出物仅为水为止。馏出物分液,有机相即为产品,约848g,纯度99%,质量收率106%,水分1.1%。
[0051]
②
化合物a3的制备
[0052]
反应瓶中,加入6l甲醇和2kg化合物a1以及10g 2,6-二叔丁基对甲苯酚,搅拌均匀,再加0.2kg湿品雷尼镍。鼓氢气,55度加热24h。gc监测原料反应完,一般会生成化合物a2和化合物a3,开始后处理。垫硅藻土过滤,滤液加6l二氯甲烷,降温0-5度,缓慢加0.5kg固体硼氢化钠,控温小于20度,加完后,30度左右反应。gc监测(反应液打稀酸里,再萃取送样)原料反应完。降温0-5度,缓慢滴加稀盐酸淬灭,淬灭过程放气放热,注意滴加速度以及控温小
于30度。浓缩,控温小于40度,浓缩出去甲醇后,加水,加二氯萃取两次,有机相水洗两次,碳酸氢钠水溶液洗一次,有机相用无水硫酸镁干燥,过滤,滤液40度以下浓缩得粗品。粗品减压蒸馏,压力约0.1,温度120℃左右,收集馏分60-80℃,得化合物a3,1.76kg,质量收率约80%,纯度99%,母液10%为高沸点物质,大部分为高聚物。
[0053]
按上述反应步骤做如下表1的对比试验,其它条件与本实施例相同。
[0054]
表1比较例的实验条件和结果
[0055][0056]
由表1和结构可知,化合物a1由于其结构特征容易聚合,而氢化时钯碳反应慢,虽然转化率较高,但聚合程度多,蒸馏出来的化合物a3收率较低,升温和换雷尼镍可以加快反应速度,减少聚合程度,提高收率,而加上阻聚剂2,6-二叔丁基对甲苯酚,可以进一步减少聚合程度,提高收率。
[0057]
③
化合物a4的制备
[0058]
反应瓶中,加入4l二氯甲烷和1kg化合物a3,恒压漏斗加1l二氯稀释的0.5l三溴化磷,降温0-5度,缓慢加三溴化磷,控温小于20度,加完后,30度左右反应16h。gc监测(反应液水洗两次再碳酸氢钠洗后送样)原料反应完,降温0-5度,缓慢加水淬灭,淬灭过程放热,注意滴加速度以及控温小于30度,水洗三次,再用碳酸氢钠溶液洗至弱碱性。有机相干燥,过滤,滤液浓缩得粗品。粗品减压蒸馏,压力约0.1,温度120度左右,收集馏分60-80度,得化合物a4,1.3g,质量收率约130%,纯度95%。
[0059]
④
化合物a5(叶立德试剂)的制备
[0060]
在1000ml三口瓶中加入500ml甲苯,然后加入100g化合物a4,1g碘化钾和100g三苯基膦。氮气保护,分水回流反应24h。tlc检测基本反应完全,浓缩除去溶剂,加入适量石油醚搅拌1h后过滤除去反应剩余的三苯基膦。得到滤饼,为叶立德试剂。
[0061]
按上述反应步骤做如下表2的对比试验,其它条件与本实施例相同。
[0062]
表2比较例的实验条件和结果
[0063]
对比例条件转化率%摩尔收率%1无溶剂50/2乙腈55/3甲苯分水80754甲苯分水,碘化钾催化9085
[0064]
由表2可知,制备wittig试剂,不同溶剂和水分对转化率有影响,其中甲苯分水的收率最高,而添加碘化钾催化,可以提高转化率,并抑制异构体的产生,提高收率。
[0065]
二、化合物vi的制备
[0066]
以麦角甾醇为原料,其路线如下所示:
[0067][0068]
上述制备路线中,具体包括以下步骤:
[0069]
s1、化合物i的制备
[0070]
植物甾醇经微生物发酵得到化合物i。
[0071]
s2、化合物ii的制备
[0072]
室温下向反应瓶中加入100g化合物1、5g dmap、100ml三乙胺及500ml dcm,氮气置换,搅拌至溶液澄清。升温回流。向体系中滴加75g对甲苯磺酰氯的dcm(200ml)溶液,约30min滴加完毕,继续回流反应1-2h。tlc监测反应至原料消失。反应完毕,将体系降温至10℃,滴加50%甲醇水溶液40ml淬灭反应,然后加入300ml水,分液,有机层用水洗涤。减压浓缩以除去大部分溶剂,加入适量甲醇,继续浓缩至dcm被完全除去。保留约100ml甲醇,降温至0-10℃,搅拌下析晶1h,抽滤,甲醇淋洗,45-50℃烘干得化合物ii。重量收率约140%,纯度大于98%。
[0073]
经检测:1h nmr(400mhz,cdcl3)δ7.75(d,j=8.1hz,2h),7.32(d,j=8.0hz,2h),5.69(s,1h),3.93(dd,j=9.2,2.9hz,1h),3.75(dd,j=9.1,6.5hz,1h),2.52
–
2.16(m,7h),2.06
–
1.88
[0074]
(m,2h),1.86
–
1.32(m,8h),1.18
–
0.78(m,13h),0.65(s,3h)。
[0075]
s3、化合物iii的制备
[0076]
室温下,搅拌下向反应瓶中加入50g化合物ii、无水甲醇250ml、2.5gpts(对甲苯磺酸)、40ml原乙酸三甲酯,保持30℃,大约3h反应完毕,tlc监测,原料反应完毕后,加入200ml丙酮、35ml水、40g四氯苯醌,搅拌下缓慢升温至40℃左右反应,tlc监测,大约3-4h反应完毕,将反应体系倒入500ml水中以析出固体,过滤,固体用200ml氯仿加热至50℃溶解,趁热过滤,滤饼用50ml氯仿加热溶解,过滤,合并有机相。有机相加入饱和亚硫酸钠水溶液(含25g亚硫酸钠)搅拌1h,静置分层,有机相减压浓缩以除去大部分溶剂,加入甲醇并继续浓缩(该操作进行3次),保留约50ml甲醇,降温至0℃析晶1h,抽滤,甲醇淋洗,45-50℃烘干得化合物3。重量收率约90%,纯度大于93%。
[0077]
经检测:1h nmr(400mhz,cdcl3)δ7.74(t,j=7.1hz,2h),7.32(d,j=7.7hz,2h),6.06(d,j=6.6hz,2h),5.62(d,j=6.5hz,1h),3.94(dd,j=9.0,2.3hz,1h),3.84
–
3.61(m,1h),2.64
–
2.46(m,1h),2.45
–
2.25(m,4h),2.13(t,j=10.1hz,1h),2.04
–
1.84(m,2h),1.82
–
1.31(m,6h),1.31
–
1.02(m,9h),1.04
–
0.89(m,3h),0.73
–
0.58(m,3h)。
[0078]
s4、化合物iv的制备
[0079]
室温下,向反应瓶中加入50g化合物iii、250ml醋酐、100ml乙酰氯,避光升温至回
流反应,约6-8h后tlc监测,原料剩余小于5%。将反应液在75℃左右减压浓缩至干,降温至室温,滴加25ml甲醇淬灭剩余醋酐加入50ml丙酮,减压浓缩以除去大部分溶剂,加入100ml丙酮并继续浓缩,保留约50ml丙酮,降温至0℃析晶1h,过滤,冰丙酮淋洗,固体45-50℃烘干得化合物vi,重量收率约90%,纯度大于95%。
[0080]
经检测:1h nmr(400mhz,cdcl3)δ7.76(d,j=7.8hz,2h),7.32(d,j=7.9hz,2h),5.73(s,1h),5.55(d,j=5.7hz,1h),5.47(s,1h),3.95(dd,j=9.2,2.3hz,1h),3.85
–
3.69(m,1h),2.54(dd,j=21.0,8.4hz,1h),2.42(s,3h),2.19
–
1.96(m,6h),1.87(dd,j=12.4,5.3hz,2h),1.77
–
1.49(m,6h),1.48
–
1.14(m,4h),1.03
–
0.91(m,6h),0.56(s,3h)。
[0081]
s5、化合物v的制备
[0082]
室温下,向反应瓶中加入3.5g无水氯化钙、20g吡啶、200ml甲醇、200ml thf,搅拌溶解;而后降温至-10~-15℃,分4批加入硼氢化钠,每批2g间隔10min,全部加完后,加入50g化合物iv,加完后保持体系温度-5~-10℃反应,大约8-10h反应完毕,tlc监测,无原料剩余;将反应液缓慢倒入500ml冰水中,边加边搅拌,待固体析出完毕搅拌20min,向体系中缓慢滴加10ml冰醋酸,抽滤,水淋洗。将固体用150ml dcm溶解,分去水层,有机相减压浓缩以除去大部分溶剂,加入甲醇并继续浓缩(该操作进行3次),最后保留甲醇约50ml,降温至0℃析晶1h,抽滤,冰甲醇淋洗,45-50℃烘干得化合物v。重量收率约70%,纯度大于95%。
[0083]
经检测:1h nmr(400mhz,cdcl3)δ7.77(d,j=8.1hz,2h),7.33(d,j=8.0hz,2h),5.54(d,j=3.7hz,1h),5.44
–
5.20(m,1h),3.95(dd,j=9.2,2.7hz,1h),3.80(dd,j=9.1,6.4hz,1h),3.67
–
3.52(m,1h),2.58
–
2.37(m,4h),2.26(t,j=12.8hz,1h),2.08
–
1.78(m,6h),1.74
–
1.14(m,11h),0.98(dd,j=11.3,6.1hz,3h),0.87(d,j=31.0hz,3h),0.59(d,j=23.9hz,3h).
[0084]
13c nmr(101mhz,cdcl3)δ144.59,140.44,140.05,133.01,129.72,127.84,119.39,116.57,75.49,70.26,53.97,51.43,46.03,42.94,40.67,38.77,38.27,36.94,36.44,31.84,27.26,22.93,21.57,20.94,16.92,16.19,11.70.(2个碳信号与其他信号重叠)
[0085]
s6、化合物vi的制备
[0086]
室温下,向反应瓶里加入50g化合物v,再加150ml无水dmso(二甲基亚砜),搅拌下加22ml的三乙胺,加热到100度反应2-3h,tlc监测原料反应完;缓慢倒入1l水中水析,抽滤,水淋洗,粗品用100ml甲醇加热打浆1-2h,降温至0℃析晶1h,抽滤,冰甲醇淋洗,45-50℃烘干得化合物vi。重量收率约60%,纯度大于95%。
[0087]
按上述反应步骤做如下表3的对比试验,其它条件与本实施例相同。
[0088]
表3比较例的实验条件和结果
[0089]
[0090][0091]
由表3可知,该反应对水分和碱的种类有要求,当水分较高时,水解杂质可达15-20%,而碱的种类不同,反应的异构体杂质差异明显,约2-30%,一般来说无机碱异构体大,有机碱异构体小,目前最优条件1.5eq三乙胺,dmso(水分《0.1%)。
[0092]
三、麦角甾醇的制备
[0093]
以上述制备的化合物vi为原料,经与上述制备的化合物a5发生wittig反应,得到麦角甾醇,其路线如下:
[0094][0095]
具体方法为:
[0096]
在1000ml三口瓶中加入500ml无水thf,加入制备得到的叶立德试剂a5,氮气保护下,0℃下滴加正丁基锂150ml,控制温度在10℃以下,然后加入50g化合物vi,缓慢升至常温反应1h。tlc监测反应。反应完毕后,滴加25ml水淬灭反应。浓缩除去thf,加250ml水,水相用石油醚(200ml*2)萃取,合并有机相,浓缩,加入甲醇并继续浓缩至小体积,搅拌降温至0~5℃,析晶1小时,过滤,得粗品。粗品加400ml无水乙醇,0.5g活性炭,30-35度搅拌脱色2h,过滤,30-35度浓缩至小体积,搅拌降温至0~5℃,析晶1小时,过滤,在30-35度真空干燥,得白色晶体麦角甾醇产品。重量收率约90%,纯度大于99%。
[0097]
经检测:1h nmr(400mhz,cdcl3)δ5.56(dd,j=10.6,7.2hz,1h),5.37(dd,j=14.1,11.3hz,1h),5.26
–
5.04(m,2h),3.71
–
3.50(m,1h),2.45(dt,j=30.1,15.1hz,1h),2.26(dd,j=25.2,12.7hz,1h),2.11
–
1.18(m,19h),1.02(t,j=7.4hz,3h),0.96
–
0.88(m,6h),0.82(dd,j=13.1,6.7hz,6h),0.63(s,3h)。
[0098]
13
cnmr(101mhz,cdcl3)δ141.35,139.77,135.56,131.98,119.59,116.28,70.47,55.74,54.56,46.26,42.83,40.79,40.41,39.09,38.38,37.04,33.09,31.99,28.28,23.00,21.11,19.95,19.64,17.60,16.28,12.05。
[0099]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0100]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护
范围。因此,本发明专利的保护范围应以所附权利要求为准。