1.本发明涉及一种一锅法制备2-氯甲基-3,4-二甲氧基吡啶盐酸盐的方法。
背景技术:
2.泮托拉唑钠是一种常用药物,由瑞士nycomedpharma公司研制开发,是一种不可逆的质子泵抑制剂,临床主要用于治疗胃溃疡和十二指肠溃疡等消化系统疾病。泮托拉唑钠是继奥美拉唑、兰索拉唑之后的新一代质子泵抑制剂,选择性更好,不良反应和药物相互作用少。2-氯甲基-3,4-二甲氧基吡啶盐酸盐为合成泮托拉唑的关键医药中间体,结构式如下:
[0003][0004]
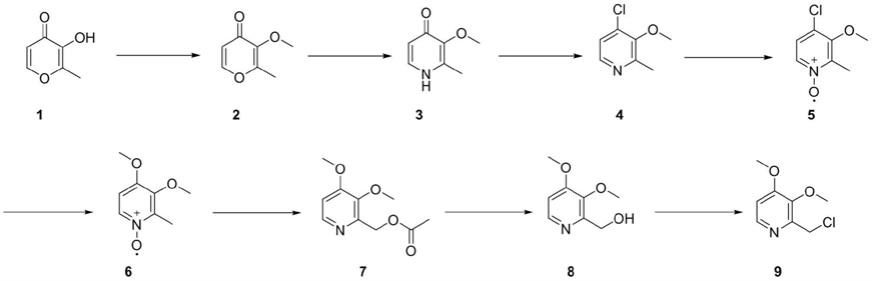
cn102301083a公开了2-氯甲基-3,4-二甲氧基吡啶盐酸盐的合成方法,采用甲基麦芽酚为原料,经过甲基化,胺化,氯化,氧化,甲氧基取代,羟甲基化,二次氯化制备而成,其反应式如下:
[0005][0006]
该制备路线中涉及的中间体3、5、8都需要进行分离,并需要重结晶纯化。操作步骤繁琐,收率不高,并且需要使用过多的溶剂进行重结晶。
[0007]
刘德龙等(徐州师范大学学报(自然科学版)vol.21,no.1,mar.,2003)也公开了上述制备路线,制备路线中需要分离出中间体2、3、5、6、7中间体。
[0008]
李荣东等(中国医药工业杂志2000年第10期475-476)同样也描述了上述制备路线,但路线整体收率低,难于工业化生产。
[0009]
在现有的制备2-氯甲基-3,4-二甲氧基吡啶盐酸盐的方法中一般都是以甲基麦芽酚为起始原料,经过甲基化,胺化,氯化,氧化,甲氧基取代,羟甲基化,二次氯化制备而成。在现有的制备路线中,多步反应中间体需要纯化结晶,步骤繁琐,大大增加了经济成本,并且总收率也不高。
技术实现要素:
[0010]
本发明公开了一种一锅法制备2-氯甲基-3,4-二甲氧基吡啶盐酸盐的方法,其生
产成本低,操作简单,收率和纯度较高,适合工业化生产,此方法在传统的工艺合成路线上简化了操作步骤,优化了反应条件,不需要分离中间体,即所谓的一锅法方法处理,从而拿到合格的产品。
[0011]
为实现上述目的,本发明提供以下技术方案:
[0012]
本发明公开了一种一锅法制备泮托拉唑中间体2-氯甲基-3,4-二甲氧基吡啶盐酸盐的方法,其特征在于,泮托拉唑中间体2-氯甲基-3,4-二甲氧基吡啶盐酸盐的反应式如下:
[0013][0014]
作为进一步地改进,本发明所述的化合物2的制备方法:将一定量的甲基麦芽酚加入到预先配置的液碱中,搅拌溶解,在10~15℃向其中滴加一定量的硫酸二甲酯,控制反应液的ph值在8-10,二甲酯滴加完成后,升温到40~45℃保温反应完全,降温,加入二氯甲烷萃取,浓缩二氯甲烷至干,直接作下一步反应,其中甲基化试剂为一氯甲烷或硫酸二甲酯。
[0015]
作为进一步地改进,本发明所述的化合物3的制备方法:加入适量溶剂,搅拌升温至40-50℃左右,通入液氨反应,反应完毕后减压浓缩,并用二氯甲烷拖带干,直接作下一步反应,反应溶剂为甲醇、异丙醇,正丁醇,异丁醇中的任意一种;或为酮类或脂类或乙腈或甲苯。
[0016]
作为进一步地改进,本发明所述的化合物4的制备方法:加入定量的二氯甲烷,35℃以下滴加三氯氧磷,滴加完毕,常压蒸馏二氯甲烷,升温70~75℃保温反应,升温并减压蒸馏三氯氧磷,将氯化液和液碱同时滴加入一定量水中,控制ph值达到8~10,分液收集下层油状物,水相加入定量的甲苯萃取,合并有机相并浓缩干,直接作下一步反应。
[0017]
作为进一步地改进,本发明所述的化合物5的制备方法:加入定量的钨酸钠或其水合物,搅拌升温至65-75℃,滴加定量的双氧水,保温4-5个小时,降温后,滴加定量的液碱,调ph值至中性,二氯甲烷萃取,浓缩二氯甲烷至干,直接作下一步反应,钨酸钠或其水合物的当量在0.1~0.5之间。
[0018]
作为进一步地改进,本发明所述的钨酸钠或其水合物的当量优选为0.1~0.2之间。
[0019]
作为进一步地改进,本发明所述的化合物6的制备方法:加入定量甲醇、以及片碱、氢氧化钾、碳酸钾、碳酸钠、碳酸铯、氟化铯、醋酸钾、磷酸钾、磷酸钠中的任意一种,升温至45-50℃左右,保温反应6-8个小时,浓缩甲醇并加入定量的水和二氯甲烷萃取,浓缩二氯甲烷至干,直接作下一步反应。
[0020]
作为进一步地改进,本发明所述的化合物8的制备方法:加入醋酸酐,升温80~85℃,保温反应完全,减压蒸馏出多余的醋酸和醋酐,加定量水、碱液进行中和水解,水解完毕
加入定量二氯甲烷萃取,直接作下一步反应。
[0021]
作为进一步地改进,本发明所述的化合物9的制备方法:二氯甲烷相降温至10℃以下,滴加氯化亚砜,保温反应完全,减压浓缩氯化亚砜和二氯甲烷,加定量无水溶剂重结晶并过滤得2-氯甲基-3,4-二甲氧基吡啶盐酸盐,加定量无水溶剂重结晶的重结晶的溶剂为乙醇,异丙醇,甲醇中的任意一种或任意几种。
[0022]
作为进一步地改进,本发明所述的反应后处理均不需要纯化分离。
[0023]
本发明的有益效果如下:
[0024]
本发明提供了一种简便的投料方式以及后处理方法,在原有的工艺上改进了操作方法以“一锅法”的方式得到2-氯甲基-3,4-二甲氧基吡啶盐酸盐,不仅大大提高了反应效率,避免了中间体的烘料等操作,简化了操作步骤,也降低了成本。一锅法工艺简单可靠,利于商业化生产实施反应总收率最高可以达到60%以上。
具体实施方式
[0025]
本发明公开了一种一锅法制备泮托拉唑中间体2-氯甲基-3,4-二甲氧基吡啶盐酸盐的方法,泮托拉唑中间体2-氯甲基-3,4-二甲氧基吡啶盐酸盐的反应路线如下:
[0026][0027]
反应路线包括以下步骤:
[0028]
将一定量的甲基麦芽酚加入到预先配置10%液碱中,搅拌溶解。在10~15℃向其中滴加一定量的硫酸二甲酯,控制反应液的ph值在9左右,二甲酯滴加完成后,升温到40~45℃保温3小时,降温到20℃以下,加入二氯甲烷萃取,浓缩二氯甲烷至干得较高纯度的化合物2,可直接用于下一步反应。如上所述反应,甲基化试剂可以为一氯甲烷,硫酸二甲酯,其中优选硫酸二甲酯。
[0029]
在化合物2中加入适量甲醇,搅拌升温至45℃左右,通入液氨反应,反应完毕后减压浓缩,并用二氯甲烷拖带至干得到化合物3。此步反应并不需要进行纯化可直接作下一步反应。如上所述化合物3制备方法,上述反应溶剂甲醇也可以为其他醇类,例如异丙醇,正丁醇,异丁醇;可以为酮类,例如丙酮,2-丁酮等;可以为脂类,例如乙酸甲酯,乙酸异丙酯等;也可以为乙腈,甲苯等。其中优先甲醇作溶剂。
[0030]
在化合物3中加入定量的二氯甲烷,35℃以下滴加三氯氧磷,滴加完毕,常压蒸馏二氯甲烷,升温70~75℃保温8h。升温并减压蒸馏三氯氧磷,将氯化液和液碱同时滴加入一定量水中,控制ph值达到8~10,分液收集下层油状物,水相加入定量的甲苯萃取,合并有机相并浓缩干得化合物4可直接用于下一步反应。
[0031]
在化合物4中加入定量的钨酸钠,搅拌升温至70℃,滴加定量的双氧水,保温4个小
时。反应完毕后降温到20℃以下,滴加定量的液碱,调ph值至中性。二氯甲烷萃取,浓缩二氯甲烷相至干,得化合物5,此步浓缩物不需要重结晶可直接作下一步反应,并不影响后面反应。一般地,钨酸钠的当量应在0.1~0.5之间,优选0.1~0.2。
[0032]
在化合物5中加入定量甲醇和片碱,升温至50℃左右,保温反应6个小时。浓缩甲醇并加入定量的水和二氯甲烷萃取,浓缩二氯甲烷至干得化合物6,如上所述反应条件,其中碱也可以为氢氧化钾,碳酸钾、碳酸钠、碳酸铯、氟化铯、醋酸钾、磷酸钾或磷酸钠。
[0033]
在化合物6中加入醋酸酐,升温80~85℃,保温6小时。减压蒸馏出多余的醋酸和醋酐,加定量水、碱液进行中和水解,水解完毕加入定量二氯甲烷萃取,如上述反应,此步反应不需要重结晶纯化可直接作下一步反应。
[0034]
将化合物7的二氯甲烷相降温至10℃以下,滴加氯化亚砜,保温4小时,减压浓缩氯化亚砜和二氯甲烷。加定量无水乙醇重结晶并过滤得2-氯甲基-3,4-二甲氧基吡啶盐酸盐,其纯度在98%以上。其中重结晶的溶剂可以为乙醇,异丙醇,甲醇等或其混合溶剂,优选乙醇。
[0035]
实施例
[0036]
实施例1:
[0037]
将50g甲基麦芽酚加入到250g预先配置10%氢氧化钠溶液中,搅拌溶解。14℃滴加65g硫酸二甲酯,控温40℃左右,控制反应液的ph值在9左右,滴加完毕,升温到45℃保温反应7小时,反应完毕降温至20℃以下,加入300ml二氯甲烷分三次萃取,合并二氯甲烷相并浓缩至干。
[0038]
加入250ml甲醇,搅拌升温至45℃左右,通入氨气反应8小时,反应完毕后减压浓缩,并用100ml二氯甲烷拖带干。
[0039]
加入150ml的二氯甲烷,35℃以下滴加约250g三氯氧磷,滴加完毕,常压蒸馏二氯甲烷,缓慢升温75℃保温16h过夜。升温并减压蒸馏三氯氧磷,将氯化液和30%氢氧化钠同时滴加入200ml水中,控制ph值达到9左右,分液并收集下层油状物,水相加入200ml的甲苯萃取,合并有机相并浓缩干。
[0040]
浓缩液加入10g二水钨酸钠,搅拌升温至70℃,滴加60g35%的双氧水,保温反应6个小时。降温到20℃以下,滴加30%氢氧化钠调节ph值至中性。300ml二氯甲烷分3次萃取,合并有机相并浓缩干。
[0041]
加入200ml甲醇和25g氢氧化钠,升温至50℃左右,保温反应16小时过夜。浓缩甲醇并加入200ml的水和300ml二氯甲烷分3次萃取,浓缩二氯甲烷至干。
[0042]
加200g醋酸酐,升温80~85℃,保温16小时过夜。减压蒸馏出多余的醋酸和醋酐,加入16%氢氧化钠120g溶液升温90℃进行中和水解,水解完毕降温并加入200ml二氯甲烷分2次萃取,合并二氯甲烷相。
[0043]
二氯甲烷相降温至10℃以下,滴加80g氯化亚砜,控温10℃以下反应4小时,减压浓缩氯化亚砜和二氯甲烷。加150ml无水乙醇重结晶,升温回流2小时后降温10℃以下搅拌6小时,过滤得2-氯甲基-3,4-二甲氧基吡啶盐酸盐烘干约43.5g,收率58.4%。
[0044]
实施例2:
[0045]
将100g甲基麦芽酚加入到500g预先配置10%氢氧化钠溶液中,搅拌溶解。12℃滴加130g硫酸二甲酯,控温40℃左右,控制反应液的ph值在9左右,滴加完毕,升温到45℃保温
反应8小时,反应完毕降温至20℃以下,加入600ml二氯甲烷分三次萃取,合并二氯甲烷相并浓缩至干。
[0046]
加入500ml甲醇,搅拌升温至45℃左右,通入氨气反应8小时,反应完毕后减压浓缩,并用200ml二氯甲烷拖带干。
[0047]
加入300ml的二氯甲烷,35℃以下滴加约500g三氯氧磷,滴加完毕,常压蒸馏二氯甲烷,缓慢升温75℃保温16h过夜。升温并减压蒸馏三氯氧磷,将氯化液和30%氢氧化钠同时滴加入400ml水中,控制ph值达到9左右,分液并收集下层油状物,水相加入400ml的甲苯萃取,合并有机相并浓缩干。
[0048]
浓缩液加入20g二水钨酸钠,搅拌升温至72℃,滴加120g35%的双氧水,保温反应6个小时。降温到20℃以下,滴加30%氢氧化钠调节ph值至中性。600ml二氯甲烷分3次萃取,合并有机相并浓缩干。
[0049]
加入400ml甲醇和50g氢氧化钠,升温至50℃左右,保温反应16小时过夜。浓缩甲醇并加入400ml的水和600ml二氯甲烷分3次萃取,浓缩二氯甲烷至干。
[0050]
加400g醋酸酐,升温85℃,保温16小时过夜。减压蒸馏出多余的醋酸和醋酐,加入16%氢氧化钠240g溶液升温90℃进行中和水解,水解完毕降温并加入400ml二氯甲烷分2次萃取,合并二氯甲烷相。
[0051]
将二氯甲烷相降温至10℃以下,滴加160g氯化亚砜,控温10℃以下反应4小时,减压浓缩氯化亚砜和二氯甲烷至干。加300ml无水乙醇重结晶,升温回流2小时后降温10℃以下搅拌10小时,过滤得2-氯甲基-3,4-二甲氧基吡啶盐酸盐烘干约87.6g,收率58.8%。
[0052]
实施例3:
[0053]
将200kg甲基麦芽酚加入到1000kg预先配置10%氢氧化钠溶液中,搅拌溶解。12℃滴加260kg硫酸二甲酯,控温40℃左右,控制反应液的ph值在9左右,滴加完毕,升温到45℃保温反应10小时,反应完毕降温至20℃以下,加入1200l二氯甲烷分三次萃取,合并二氯甲烷相并浓缩至干。
[0054]
加入1000l甲醇,搅拌升温至45℃左右,通入氨气反应12小时,反应完毕后减压浓缩,并用400l二氯甲烷拖带干。
[0055]
加入600l的二氯甲烷,35℃以下滴加约1000kg三氯氧磷,滴加完毕,常压蒸馏二氯甲烷,缓慢升温75℃保温20h过夜。升温并减压蒸馏三氯氧磷,将氯化液和30%氢氧化钠同时滴加入800l水中,控制ph值达到9左右,分液并收集下层油状物,水相加入800l的甲苯萃取,合并有机相并浓缩干。
[0056]
浓缩液加入40kg二水钨酸钠,搅拌升温至72℃,滴加240kg35%的双氧水,保温反应6个小时。降温到20℃以下,滴加30%氢氧化钠调节ph值至中性。1200l二氯甲烷分3次萃取,合并有机相并浓缩干。
[0057]
加入800l甲醇和100kg氢氧化钠,升温至50℃左右,保温反应16小时过夜。浓缩甲醇并加入800l的水和600l二氯甲烷分3次萃取,浓缩二氯甲烷至干。
[0058]
加800kg醋酸酐,升温85℃,保温16小时过夜。减压蒸馏出多余的醋酸和醋酐,加入16%氢氧化钠480kg溶液升温90℃进行中和水解,水解完毕降温并加入800l二氯甲烷分2次萃取,合并二氯甲烷相。
[0059]
将二氯甲烷相降温至10℃以下,滴加320kg氯化亚砜,控温10℃以下反应4小时,减
压浓缩氯化亚砜和二氯甲烷至干。加500l无水乙醇重结晶,升温回流2小时后降温10℃以下搅拌10小时,过滤得2-氯甲基-3,4-二甲氧基吡啶盐酸盐烘干约185kg,收率62.2%。
[0060]
本领域普通技术人员可以理解,以上所述仅为发明的优选实例而已,并不用于限制发明,尽管参照前述实例对发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实例记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在发明的精神和原则之内,所做的修改、等同替换等均应包含在发明的保护范围之内。