1.本发明属于医药领域,具体地说,是一种合成萘喹酯的新方法。
背景技术:
2.萘喹酯又名甲氧苄喹酯、苄氧喹甲酯,化学名4-羟基-6-丁基-7-苄氧基喹啉-3-羧酸甲酯(6-butyl-1,4-dihydro-4-oxo-7-(phenyl methoxy)-3-quinolinecarboxylic acid methyl ester),分子式c
22h23
no4,为新型喹啉类广谱抗球虫药物。在球虫的无性繁殖阶段发挥作用,进入子孢子细胞后,通过干扰dna合成而阻止其发育。在球虫生活史的早期即开始发挥作用,从而避免家禽肠道受到损害。萘喹酯1的结构式,如下所示:
[0003][0004]
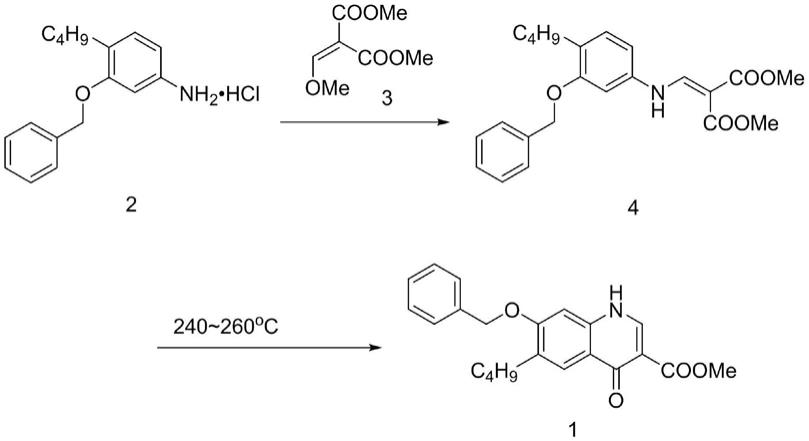
在现有技术中关于萘喹酯1的合成,其中一种方法是通过化合物2与化合物3缩合反应后生成化合物4,然后化合物4在适当的溶剂中高温环合而成,反应方程式如下所示:
[0005][0006]
在上述合成路线中,第二步反应温度高且反应时间短,反应时间往往不长于10分钟,反应时间延长奈喹酯降解变质,进而影响到后续的纯化。因此,用此路线合成奈喹酯收率较低,产品难以纯化,成本高。另外,过短的反应时间无法采用传统的间歇式反应釜进行操作,给工业化生产带来了巨大的困难。
技术实现要素:
[0007]
本发明所要解决的技术问题在于提供一种合成萘喹酯的新方法,以克服传统合成路线的缺陷及收率低和成本高等劣势。
[0008]
为解决上述技术问题,本发明采用的技术方案如下:
[0009]
本发明公开了一种合成萘喹酯的新方法,反应式如下:
[0010][0011]
包括以下步骤:
[0012]
1)将化合物2用无机碱或有机碱游离,有机溶剂萃取,浓缩溶剂后,加入化合物5加热保温反应,反应完毕后除去溶剂,得6粗品,直接用于步骤2);
[0013]
2)将步骤1)得到的粗品6与溶剂混合中,升温反应,反应完毕后降温,加入适当的溶剂混合搅拌后,过滤、洗涤得化合物7;
[0014]
3)将步骤2)得到的化合物7用碱水解,然后酸化得化合物8;
[0015]
4)将步骤3)得到的化合物8甲酯化,甲酯化通常采用甲醇/二氯亚砜体系,后处理得到奈喹酯粗品,再重结晶得产品1。
[0016]
作为进一步地改进,本发明所用无机碱为氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾中的任意一种。
[0017]
作为进一步地改进,本发明所述的步骤1)中,所用溶剂为乙酸乙酯、乙酸异丙酯、甲苯中的任意一种。
[0018]
作为进一步地改进,本发明所述的步骤1)中,反应温度为60~120℃。
[0019]
作为进一步地改进,本发明所述的步骤2)中,所述溶剂是二苯醚,联苯,甘油,聚乙二醇二甲醚,dowtherm a中的任意一种。
[0020]
作为进一步地改进,本发明所述的步骤2)中,化合物6与溶剂的质量比为1:2~10。
[0021]
作为进一步地改进,本发明所述的步骤2)中,反应温度为240~260℃。
[0022]
作为进一步地改进,本发明所述的步骤2)中,所述的反应时间为5~30min。
[0023]
作为进一步地改进,本发明所述的步骤2)中,因反应时间短,化合物6反应不完全,将过滤后将母液再次升温反应,反复多次直至化合物6反应完全。
[0024]
作为进一步地改进,本发明所述的步骤3)中,所用碱为氢氧化钠、氢氧化钾中的任意一种,所用酸为盐酸、磷酸、硫酸中的任意一种。
[0025]
本发明与现有技术相比,优势在于:
[0026]
(1)采用价格较低的化合物5,相比化合物3成本优势明显;
[0027]
(2)本发明可有效提高转化率,易操作。
[0028]
(3)本发明的新方法产品产率最高可达78%。
具体实施方式
[0029]
本发明公开了—种合成萘喹酯的新方法,反应式如下:
[0030][0031]
同时,我们还公开了具体反应步骤,如下:
[0032]
(1)将化合物2用无机碱或有机碱游离,有机溶剂萃取,浓缩溶剂后,加入化合物5加热保温反应,反应完毕后除去溶剂,得6粗品,直接用于步骤(2);
[0033]
(2)将步骤(1)得到的粗品6与溶剂混合中,升温反应,反应完毕后降温,加入适当的溶剂混合搅拌后,过滤、洗涤得化合物7;
[0034]
(3)将步骤(2)得到的化合物7用碱水解,然后酸化得化合物8;
[0035]
(4)将步骤(3)得到的化合物8甲酯化,甲酯化通常采用甲醇/二氯亚砜体系,后处理得到奈喹酯粗品,再重结晶得产品1。
[0036]
步骤(1)中,所用无机碱为氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾,优选为氢氧化钠;
[0037]
步骤(1)中,所用溶剂为乙酸乙酯、乙酸异丙酯、甲苯,优选为乙酸乙酯;特别强调在这一步骤中,可在萃取后直接除去溶剂得到游离胺粗品,然后与化合物5进行无溶剂反应;
[0038]
步骤(1)中,反应温度为60~120℃,优选80~100℃;
[0039]
步骤(2)中,所述溶剂是二苯醚,联苯,甘油,聚乙二醇二甲醚,dowtherma等,优选二苯醚;
[0040]
步骤(2)中,化合物6与溶剂的质量比为1:2~10,优选1:2~5;
[0041]
步骤(2)中,反应温度为240~260℃,优选250~255℃;
[0042]
步骤(2)中,反应时间为5~30min,优选10~15min;
[0043]
步骤(2)中,因反应时间短,化合物6反应不完全,可将过滤后将母液再次升温反应,反复多次直至化合物6反应完全;
[0044]
步骤(3)中,所用碱为氢氧化钠、氢氧化钾,优选为氢氧化钠;
[0045]
步骤(3)中,所用酸为盐酸、磷酸、硫酸,优选为盐酸。
[0046]
实施例
[0047]
下面通过具体实施例对本发明的技术方案作进一步地说明,根据下述实施例,可以更好地理解本发明。然而,本领域的技术人员容易理解,实施例所描述的内容仅用于说明本发明,而不应当也不会限制权利要求书中所详细描述的本发明。
[0048]
以下实施例所使用的化合物2为自制,化合物5为rg,其他试剂均为ar。
[0049]
实施例1:化合物7的制备
[0050][0051]
取24g氢氧化钠,溶于600g水中,加入540g乙酸乙酯,搅拌,再分批加入149.5g化合物2,搅拌30min后静置分层,有机层用10g无水硫酸钠脱水后减压除去乙酸乙酯。在游离胺粗品中加入化合物5,升温至80℃,保温反应2h,反应完毕减压除去乙醇,得化合物6。
[0052]
将上诉化合物6加入380g二苯醚中,迅速升温至250~255℃,保温反应10~15min后降温至40℃,加入380g石油醚并搅散固体,过滤,滤饼用少量石油醚洗涤后烘干;滤液先升温蒸除石油醚,再迅速升温至250~255℃,保温反应10~15min后降温至40℃,加入380g石油醚并搅散固体,过滤,滤饼用少量石油醚洗涤后烘干,如此反应3次,化合物6基本反应完毕,得化合物7共161.8g,收率85.3%。
[0053]
实施例2:化合物8的制备
[0054][0055]
取15.5g氢氧化钠,溶于140g水中,依次加入100g乙醇和49g化合物7,升温至回流,保温反应3h,反应完毕后滴加30%盐酸,析出大量固体,降温至室温,搅拌30min,过滤,滤饼用水洗涤后烘干,得44g化合物8,收率97%。
[0056]
实施例3:化合物1的制备
[0057][0058]
取44g化合物8,将其加入760g甲醇中,降温至0~5℃,滴加135.6g二氯亚砜后搅拌30min,升温至回流,保温反应24h,反应完毕降温至室温,将其倒入1000g 5%碳酸氢钠溶液中淬灭,析出大量固体,搅拌30min后过滤,滤饼用水洗涤后烘干,得化合物1粗品,再用dmf重结晶,得44.9g化合物1,收率95.2%。
[0059]
实施例4:化合物7的制备
[0060][0061]
取34.6g碳酸钾,溶于600g水中,加入520g甲苯,搅拌,再分批加入149.5g化合物2,搅拌30min后静置分层,有机层用10g无水硫酸钠脱水后加入化合物5,升温至100℃,保温反应2h,反应完毕减压除去甲苯,得化合物6。
[0062]
将上诉化合物6加入380g二苯醚中,迅速升温至250~255℃,保温反应10~15min后降温至40℃,加入380g石油醚并搅散固体,过滤,滤饼用少量石油醚洗涤后烘干;滤液先升温蒸除石油醚,再迅速升温至250~255℃,保温反应10~15min后降温至40℃,加入380g石油醚并搅散固体,过滤,滤饼用少量石油醚洗涤后烘干,如此反应3次,化合物6基本反应完毕,得化合物7共160.8g,收率84.8%。
[0063]
实施例5:化合物8的制备
[0064][0065]
取21.7g氢氧化钾,溶于140g水中,依次加入100g乙醇和49g化合物7,升温至回流,保温反应3h,反应完毕后滴加78.4g50%硫酸,析出大量固体,降温至室温,搅拌30min,过滤,滤饼用水洗涤后烘干,得44.1g化合物8,收率97.3%。
[0066]
本领域普通技术人员可以理解,以上所述仅为发明的优选实例而已,并不用于限制发明,尽管参照前述实例对发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实例记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在发明的精神和原则之内,所做的修改、等同替换等均应包含在发明的保护范围之内。