1.本发明属于有机化合物技术领域,具体涉及一种吡啶3-胺衍生物及其制备方法和应用。
背景技术:
2.高尿酸血症,是指在正常嘌呤饮食状态下,非同日两次空腹血尿酸水平男性高于420μmol/l,女性高于360μmol/l。高尿酸血症与高血压、糖尿病、肾脏疾病、心血管疾病等的发生密切相关。临床上高尿酸血症最普遍的表现为痛风。痛风的形成原因是尿酸结晶在关节处沉积,引起关节处严重的疼痛、红肿以及炎症。它是男性最常见的慢性炎症性关节炎,也是越来越多的女性关节炎的病因。世界范围内的流行病学表明,发达国家和发展中国家的发病率和患病率都在增加。
3.高尿酸血症的形成原因是体内尿酸代谢紊乱:包括尿酸的生成增加以及肾脏排泄尿酸减少。尿酸的形成离不开黄嘌呤氧化酶(xanthine oxidoreductase),因此治疗高尿酸血症的药物根据它们的作用机制分为两种,黄嘌呤氧化酶抑制剂(xois)和促尿酸排泄药。事实上,超过90%的病人的高尿酸症发生原因是尿酸重吸收过多,造成排泄障碍,而尿酸盐转运蛋白1(urat1)承担了接近90%的尿酸重吸收,因此urat1是治疗高尿酸症以及痛风的重要靶点。urat1抑制剂被认为是治疗高尿酸血症的一种有效的促尿酸排泄药。
4.目前应用于临床的促尿酸排泄药,主要包括水杨酸类药物、丙磺舒、磺吡酮、苯溴马隆等。其中,丙磺舒与苯溴马隆均能抑制urat1,降低肾小管对尿酸重吸收,促进尿酸排泄,但丙磺舒易引起胃肠道不良反应如恶心或呕吐等,对造血系统偶尔会引起白细胞减少、骨髓抑制及肝坏死等不良反应;苯溴马隆则有可能引起严重的肝损伤。verinurad是阿斯利康公司开发的一种新的urat1抑制剂,通过对lesinurad进行结构修饰,用吡啶替代三氮唑。在体外研究中,verinurad显示出苯溴马隆的3倍效力,并且目前正处于ii期临床试验,用于治疗痛风和无症状高尿酸血症。因此,基于verinurad设计出药效更强,毒性更低的urat1抑制剂具有重要意义。
技术实现要素:
5.本发明旨在至少解决现有技术中存在的上述技术问题之一。为此,本发明提供了一种吡啶3-胺衍生物,该吡啶3-胺衍生物具有urat1抑制活性。
6.本发明还提供了一种吡啶3-胺衍生物的制备方法。
7.本发明还提供了一种吡啶3-胺衍生物的应用。
8.本发明的第一方面提供了一种吡啶3-胺衍生物,具有如下所示的结构:
[0009][0010]
其中,y选自n或o;
[0011]
r1选自h或c1~c4烷基;
[0012]
r2选自-cn或-conh2;
[0013]
x选自1,3-亚丙基、1,4-亚丁基、1,5-亚戊基、1,6-亚己基、1-甲基-1,1-亚乙基、2-亚甲基苯基、3-亚甲基苯基、4-亚甲基苯基、3-甲氧基-4-亚甲基苯基或4-亚甲基-6
’‑
联苯基。
[0014]
本发明关于吡啶3-胺衍生物的技术方案中的一个技术方案,至少具有以下有益效果:
[0015]
本发明的吡啶-3-胺衍生物,具有良好的urat1抑制活性,可用于制备促尿酸排泄药物。
[0016]
本发明的吡啶-3-胺衍生物,还可以进一步研制开发成为治疗高尿酸血症的新型药物。
[0017]
根据本发明的一些实施方式,y为n,r1为-ch3、r2为-cn时,所述吡啶3-胺衍生物具有如式i所示的结构:
[0018][0019]
根据本发明的一些实施方式,式i中,x为1,6-亚己基时,吡啶3-胺衍生物的结构为:
[0020][0021]
根据本发明的一些实施方式,式i中,x为1,5-亚戊基时,吡啶3-胺衍生物的结构为:
[0022][0023]
根据本发明的一些实施方式,式i中,x为1,4-亚丁基时,吡啶3-胺衍生物的结构为:
[0024][0025]
根据本发明的一些实施方式,式i中,x为1,3-亚丙基时,吡啶3-胺衍生物的结构为:
[0026][0027]
根据本发明的一些实施方式,式i中,x为1-甲基-1,1-亚乙基时,吡啶3-胺衍生物的结构为:
[0028][0029]
根据本发明的一些实施方式,式i中,x为3-亚甲基苯基时,吡啶3-胺衍生物的结构为:
[0030][0031]
根据本发明的一些实施方式,式i中,x为2-亚甲基苯基时,吡啶3-胺衍生物的结构为:
[0032][0033]
根据本发明的一些实施方式,式i中,x为4-亚甲基苯基时,吡啶3-胺衍生物的结构为:
[0034][0035]
根据本发明的一些实施方式,式i中,x为3-甲氧基-4-亚甲基苯基时,吡啶3-胺衍生物的结构为:
[0036][0037]
根据本发明的一些实施方式,式i中,x为4-亚甲基-6
’‑
联苯基时,吡啶3-胺衍生物的结构为:
[0038][0039]
根据本发明的一些实施方式,r1为h、r2为-cn时,所述吡啶3-胺衍生物具有如式ii所示的结构:
[0040]
[0041]
根据本发明的一些实施方式,式ii中,x为1,6-亚己基时,吡啶3-胺衍生物的结构为:
[0042][0043]
根据本发明的一些实施方式,式ii中,x为1,5-亚戊基时,吡啶3-胺衍生物的结构为:
[0044][0045]
根据本发明的一些实施方式,式ii中,x为1,4-亚丁基时,吡啶3-胺衍生物的结构为:
[0046][0047]
根据本发明的一些实施方式,式ii中,x为1,3-亚丙基时,吡啶3-胺衍生物的结构为:
[0048][0049]
根据本发明的一些实施方式,式ii中,x为3-亚甲基苯基时,吡啶3-胺衍生物的结构为:
[0050][0051]
根据本发明的一些实施方式,式ii中,x为2-亚甲基苯基时,吡啶3-胺衍生物的结构为:
[0052][0053]
根据本发明的一些实施方式,式ii中,x为4-亚甲基苯基时,吡啶3-胺衍生物的结构为:
[0054][0055]
根据本发明的一些实施方式,式ii中,x为3-甲氧基-4-亚甲基苯基时,吡啶3-胺衍生物的结构为:
[0056][0057]
根据本发明的一些实施方式,式ii中,x为4-亚甲基-6
’‑
联苯基时,吡啶3-胺衍生物的结构为:
[0058]
为
[0059]
根据本发明的一些实施方式,r1为-ch3、r2为-conh2时,所述吡啶3-胺衍生物具有如式iii所示的结构:
[0060][0061]
根据本发明的一些实施方式,式iii中,x为2-亚甲基苯基时,吡啶3-胺衍生物的结
构为:
[0062][0063]
根据本发明的一些实施方式,式iii中,x为3-甲氧基-4-亚甲基苯基时,吡啶3-胺衍生物的结构为:
[0064][0065]
根据本发明的一些实施方式,r1为h、r2为-conh2时,所述吡啶3-胺衍生物具有如式iv所示的结构:
[0066][0067]
根据本发明的一些实施方式,式iv中,x为3-亚甲基苯基时,吡啶3-胺衍生物的结构为:
[0068][0069]
根据本发明的一些实施方式,式iv中,x为2-亚甲基苯基时,吡啶3-胺衍生物的结构为:
[0070][0071]
根据本发明的一些实施方式,y为o,r1为-ch3、r2为-cn时,所述吡啶3-胺衍生物具有如式v所示的结构:
[0072][0073]
本发明的第二方面提供了吡啶3-胺衍生物的制备方法,括以下步骤:
[0074]
s1:以4-溴-8-萘甲腈为原料进行buchwald反应;
[0075]
s2:将步骤s1的产物与硫化钠反应生成硫酚;
[0076]
s3:将步骤s2制备得到的硫酚与溴代酯发生亲电取代连接侧链,水解得到羧酸;
[0077]
s4:将步骤s3得到的羧酸制备成钠盐。
[0078]
根据本发明的一些实施方式,吡啶3-胺衍生物的制备方法,包括以下步骤:以4-溴-8-萘甲腈为原料,经过buchwald反应合成吡啶-3-胺衍生物,再直接与硫化钠反应生成硫酚或取代n上h后再制备硫酚,硫酚与溴代酯发生亲电取代连接侧链,再水解得到羧酸,最后将羧酸制备成钠盐得到产物。
[0079]
根据本发明的一些实施方式,式i结构的吡啶3-胺衍生物的反应步骤为:
[0080][0081]
根据本发明的一些实施方式,式ii结构的吡啶3-胺衍生物的反应步骤为:
[0082][0083]
根据本发明的一些实施方式,式iii结构的吡啶3-胺衍生物的反应步骤为:
[0084][0085]
根据本发明的一些实施方式,式iv结构的吡啶3-胺衍生物的反应步骤为:
[0086][0087]
根据本发明的一些实施方式,式v结构的吡啶3-羟基衍生物的反应步骤为:
[0088][0089]
本发明的第三方面提供了上述的吡啶3-胺衍生物或其药学上可接受的盐在制备
urat1抑制剂中的应用。
[0090]
本发明的第四方面提供了上述的吡啶3-胺衍生物或其药学上可接受的盐在制备治疗高尿酸血症药物或痛风药物中的应用。
[0091]
本发明的第五方面提供了上述的一种药物组合物,含有如上所述的吡啶3-胺衍生物或其药学上可接受的盐。
[0092]
根据本发明的一些实施方式,药学上可接受的盐包括但不仅限于无机酸盐,有机酸盐,烷基磺酸盐和芳基磺酸盐中的至少一种。
[0093]
根据本发明的一些实施方式,所述无机酸盐包括但不仅限于盐酸盐、氢溴酸盐、硝酸盐、硫酸盐和磷酸盐中的至少一种;优选地,所述有机酸盐包括但不仅限于甲酸盐、乙酸盐、丙酸盐、苯甲酸盐、马来酸盐、富马酸盐、琥珀酸盐、酒石酸盐和柠檬酸盐中的至少一种。
[0094]
根据本发明的一些实施方式,所述烷基磺酸盐包括但不仅限于甲基磺酸盐和乙基磺酸盐中的至少一种。
[0095]
根据本发明的一些实施方式,所述芳基磺酸盐包括但不仅限于苯磺酸盐和对甲苯磺酸盐中的至少一种。
[0096]
根据本发明的一些实施方式,所述药物组合物包括固体口服制剂、液体口服制剂或注射剂。
[0097]
根据本发明的一些实施方式,所述固体口服制剂的剂型为胶囊或片剂。
[0098]
根据本发明的一些实施方式,所述口服制剂的剂量约为1-20mg/kg体重。
附图说明
[0099]
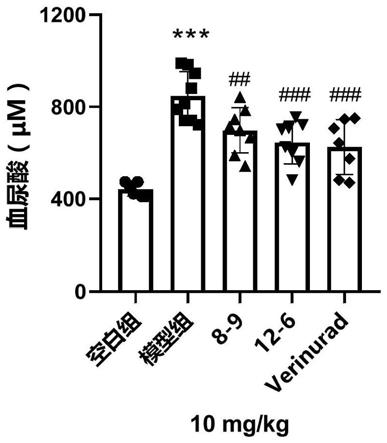
图1是化合物8-9与12-6在高尿酸血症小鼠体内降低血清尿酸作用(n=8)比较结果。
[0100]
图2是化合物8-9与12-6在高尿酸血症小鼠体内促尿酸排泄作用(n=8)比较结果。
具体实施方式
[0101]
以下是本发明的具体实施例,并结合实施例对本发明的技术方案作进一步的描述,但本发明并不限于这些实施例。
[0102]
实施例1
[0103]
本实施例制备了x为1,6-亚己基时的吡啶3-胺衍生物。式8-1的结构为:
[0104][0105]
制备方法由以下步骤组成:
[0106]
步骤一:化合物3的制备
[0107][0108]
250ml三颈瓶中加入4-溴萘甲腈(2.00g,8.62mmol),3-氨基-4氯吡啶(1.11g,8.62mmol),1,4-二氧六环(50毫升),cs2co3(8.42g,25.85mmol)、pd2(dba)3(98.64mg,0.11mmol)和binap(160.98mg,0.26mmol),然后反应混合物在氮气氛围中,102℃下搅拌过夜。tlc显示反应完成后,冷却至室温后,减压蒸发出除去溶剂,并向残渣加水(50ml),再用乙酸乙酯(200ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,硅胶柱层析纯化。得到的目标产物为黄色固体,收率为56%。
[0109]1h nmr(400mhz,cdcl3)δ8.68(s,1h),8.31(d,j=8.3hz,1h),8.12(d,j=8.4hz,1h),7.85(d,j=8.0hz,1h),7.78(t,j=7.6hz,1h),7.70(t,j=7.7hz,1h),7.47(s,1h),7.29(d,j=4.2hz,1h),6.62(s,1h)。
[0110]
步骤二:化合物4的制备
[0111][0112]
在中间体3(1.06g,3.79mmol)的四氢呋喃溶液中,分批加入氢化钠(606mg,15.16mmol,60%矿物油)。在0℃搅拌半小时后加入碘甲烷(1.08g,7.58mmol),室温搅拌过夜。反应完成后(通过tlc监测),水浴中旋转蒸发除去溶剂。再加入50ml水,用乙酸乙酯(150ml
×
3)萃取,合并有机相,加无水硫酸钠干燥,用硅胶柱层析进行纯化。得到的目标产物为淡棕色固体,产率为75%。1h nmr(400mhz,dmso)δ8.31(d,j=5.2hz,1h),8.28(s,1h),8.11(t,j=8.3hz,2h),7.81(d,j=8.5hz,1h),7.73(t,j=7.6hz,1h),7.61(d,j=5.2hz,1h),7.57(t,j=7.7hz,1h),7.32(d,j=8.0hz,1h),3.41(s,3h)。
[0113]
步骤三:化合物5的制备
[0114][0115]
在100ml烧瓶中,中间体5(1.12g,3.81mmol)溶在dmf(15ml)中,向溶液中加入九水合硫化钠(2.75g,11.44mmol),在氮气保护下,120℃下搅拌,直到tlc显示反应完成(通常在2小时内)。加入50ml的水,滴加1n的稀盐酸,边加入边搅拌,调至ph 3-4。析出的沉淀物通过减压过滤收集,得到目标黄色化合物(产率:95%)。1h nmr(400mhz,dmso)δ12.46(s,1h),8.09(d,j=8.1hz,1h),8.00(d,j=8.1hz,1h),7.75(d,j=8.5hz,1h),7.64(t,j=7.1hz,1h),7.48(s,2h),7.41(d,j=7.3hz,1h),7.32(d,j=8.1hz,1h),7.19(s,1h),3.30(s,3h)。
[0116]
步骤四:化合物6-1的制备
[0117][0118]
将硫酚5(150mg,0.52mmol)和cs2co3(335.5mg,1.04mmol)加入到5ml的dmf中,常温下搅拌10min后,加入7-溴庚酸甲酯(0.62mmol)进行反应,50℃下搅拌过夜。tlc监测反应结束后,向反应液中加入20ml水,用乙酸乙酯(100ml
×
3)萃取,过柱分离纯化得到6-1。1h nmr(400mhz,dmso)δ8.24(d,j=5.3hz,1h),8.12(d,j=8.0hz,1h),8.05(d,j=8.2hz,1h),7.76(s,1h),7.69
–
7.63(m,2h),7.45
–
7.36(m,3h),3.58(s,3h),3.30(s,3h),3.11(t,j=7.2hz,2h),2.29(t,j=7.3hz,2h),1.67(dt,j=14.8,7.3hz,2h),1.52(dt,j=15.0,7.4hz,3h),1.47
–
1.38(m,2h),1.35
–
1.28(m,2h)。
[0119]
步骤五:化合物7-1的制备
[0120][0121]
6-1(0.5g,1.15mmol)溶于甲醇(10ml)中,加入lioh(0.138g,5.77mmol)水溶液1ml,反应液在50℃搅拌过夜。然后减压蒸发除去甲醇,并向残渣中加入10ml冰水。然后用稀释的盐酸(1n)将溶液酸化至ph 3-4,溶液中生成白色固体,减压过滤取滤渣,得到目标产物7-1。1h nmr(400mhz,dmso)δ8.23(d,j=5.3hz,1h),8.12(d,j=8.0hz,1h),8.04(d,j=8.3hz,1h),7.75(s,1h),7.66(t,j=7.7hz,2h),7.47
–
7.36(m,3h),3.29(s,3h),3.12(t,j=7.2hz,2h),2.20(t,j=7.3hz,2h),1.67(dt,j=14.7,7.3hz,2h),1.52
–
1.40(m,4h),1.32(dd,j=14.7,8.0hz,3h)。
[0122]
步骤六:化合物8-1的制备
[0123][0124]
7-1(准确称量到四位小数,0.5mmol)溶于甲醇(5ml)加入氢氧化钠(0.5mmol)水溶液0.5ml,反应液在室温下搅拌直到溶液变澄清(通常需要1h),过滤除去不溶物,滤液减压蒸发除去溶剂,加入二氯甲烷(10ml
×
3)共沸除去甲醇。残渣用二氯甲烷洗涤,干燥后得到产物8-1。1h nmr(400mhz,dmso)δ8.22(d,j=5.2hz,1h),8.11(d,j=8.0hz,1h),8.03(d,j=8.3hz,1h),7.72(s,1h),7.65(t,j=8.9hz,2h),7.44
–
7.36(m,3h),3.28(s,3h),3.10(t,j=7.1hz,2h),1.89(t,j=7.3hz,2h),1.66(dt,j=14.0,7.0hz,2h),1.42(dt,j=16.0,
8.1hz,4h),1.32
–
1.25(m,2h)。
[0125]
实施例2
[0126]
本实施例制备了x为1,5-亚己基时的吡啶3-胺衍生物。式8-2的结构为:
[0127][0128]
(8-2)化合物的制备方法与8-1制备方法相同。
[0129]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)δ8.20(d,j=5.0hz,1h),8.08(d,j=8.3hz,1h),8.00(d,j=7.8hz,1h),7.73(d,j=8.6hz,1h),7.63(dd,j=17.7,10.2hz,2h),7.47(d,j=5.1hz,1h),7.38(dd,j=14.8,7.7hz,2h),3.35(s,3h),3.15(t,j=7.1hz,2h),2.22(t,j=7.3hz,2h),1.81(dd,j=19.0,11.9hz,2h),1.69(dt,j=14.2,7.1hz,2h),1.62
–
1.51(m,2h)。
[0130]
实施例3
[0131]
本实施例制备了x为1,4-亚己基时的吡啶3-胺衍生物。式8-3的结构为:
[0132][0133]
(8-3)化合物的制备方法与8-1制备方法相同。将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)δ8.20(d,j=5.3hz,1h),8.10(d,j=8.3hz,1h),8.01(t,j=9.5hz,1h),7.75(d,j=8.6hz,1h),7.70
–
7.59(m,2h),7.50(d,j=5.4hz,1h),7.41(dd,j=13.7,7.9hz,2h),3.37(s,3h),3.18(s,2h),2.27(s,2h),1.84(s,4h)。
[0134]
实施例4(制备8-4)
[0135]
结构式为化合物的制备方法同8-1。
[0136]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)δ8.19(d,j=5.3hz,1h),8.09(d,j=8.3hz,1h),8.00(d,j=8.0hz,1h),7.75(d,j=8.6hz,1h),7.68
–
7.54(m,3h),7.39(t,j=8.4hz,2h),3.36(s,3h),3.18(t,j=7.5hz,2h),2.39(t,j=7.1hz,2h),2.12
–
2.00(m,2h)。
[0137]
实施例5(制备8-5)
[0138]
结构式为化合物的制备方法同8-1。
[0139]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)δ8.12
–
8.06(m,2h),8.01(d,j=8.0hz,1h),7.79(d,j=8.6hz,1h),7.70(d,j=5.5hz,1h),7.65
–
7.57(m,2h),7.40(dd,j=18.3,8.1hz,2h),3.37(s,3h),1.78
–
1.70(m,6h)。
[0140]
实施例6(制备8-6)
[0141]
结构式为化合物的制备方法同8-1。
[0142]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)δ8.17
–
8.06(m,3h),7.97(d,j=8.0hz,1h),7.89(d,j=7.7hz,1h),7.71(d,j=8.6hz,1h),7.65(s,1h),7.59(t,j=7.6hz,1h),7.54(t,j=5.2hz,2h),7.42
–
7.31(m,3h),4.45(s,2h),3.35(d,j=4.4hz,3h)。
[0143]
实施例7(制备8-7)
[0144]
结构式为化合物的制备方法同8-1。
[0145]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,d2o)δ7.99(d,j=5.1hz,1h),7.61(dd,j=16.0,7.9hz,2h),7.54
–
7.46(m,2h),7.35
–
7.25(m,2h),7.19(d,j=4.5hz,2h),7.11(d,j=6.6hz,1h),7.05(d,j=7.4hz,1h),6.93(dd,j=18.4,8.2hz,2h),4.44(s,2h),2.94(s,3h)。
[0146]
实施例8(制备8-8)
[0147]
结构式为化合物的制备方法同8-1。
[0148]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)
δ8.11(dd,j=11.9,6.9hz,2h),7.98(d,j=8.0hz,1h),7.93(d,j=8.1hz,2h),7.70(dd,j=14.5,5.9hz,2h),7.60(t,j=7.4hz,1h),7.53
–
7.44(m,3h),7.38(d,j=8.0hz,1h),7.35
–
7.30(m,1h),4.44(s,2h),3.36(s,3h)。
[0149]
实施例9(制备8-9)
[0150]
结构式为化合物的制备方法同8-1。
[0151]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,d2o)δ7.86(s,1h),7.62(d,j=7.9hz,1h),7.55(s,1h),7.46(d,j=7.4hz,1h),7.36(d,j=11.4hz,2h),7.31(d,j=7.6hz,1h),7.14(s,1h),6.97(d,j=8.8hz,3h),6.72(d,j=7.4hz,1h),3.72(s,2h),3.59(s,3h),2.87(s,3h)
[0152]
实施例10(制备8-10)
[0153]
结构式为化合物的制备方法同8-1。
[0154]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)δ8.15(d,j=5.4hz,1h),8.10(d,j=8.1hz,1h),8.00
–
7.96(m,1h),7.77
–
7.73(m,1h),7.68(s,1h),7.63
–
7.58(m,1h),7.56(s,1h),7.54(d,j=3.2hz,2h),7.50(s,1h),7.49
–
7.47(m,2h),7.38(t,j=8.1hz,2h),7.32(t,j=3.7hz,3h),4.44(s,2h),3.37(s,3h)。
[0155]
实施例11(制备12-1)
[0156]
结构式为化合物的制备方法由以下步骤组成:
[0157]
步骤一:化合物的制备。
[0158]
步骤二:化合物的制备同化合物5。
[0159]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为1h nmr(400mhz,dmso)δ13.00(s,1h),9.50(s,1h),8.35(d,j=8.2hz,1h),8.20(s,1h),8.12(d,j=8.2hz,1h),8.06(d,j=8.1hz,1h),7.83(dt,j=15.2,7.0hz,2h),7.65(d,j=6.0hz,1h),7.58
–
7.50(m,2h)。
[0160]
步骤三:化合物的制备同化合物6-1。
[0161]
步骤四:化合物的制备同化合物7-1。
[0162]
步骤五:化合物的制备同化合物8-1。将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)δ8.55(s,1h),8.35(dd,j=12.0,7.0hz,2h),8.27(s,1h),8.11(d,j=8.3hz,1h),7.77
–
7.70(m,2h),7.68
–
7.62(m,1h),7.48(d,j=5.4hz,1h),6.34(d,j=8.2hz,1h),3.00(dd,j=9.1,5.4hz,2h),2.13(t,j=7.5hz,2h),1.67(dt,j=14.9,7.3hz,2h),1.56(dd,j=15.0,7.5hz,2h),1.44(dd,j=14.8,7.3hz,2h),1.38
–
1.32(m,2h)。
[0163]
实施例12(制备12-2)
[0164]
结构式为化合物的制备方法同2-1。
[0165]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)δ8.55(s,1h),8.35(dd,j=12.0,7.0hz,2h),8.27(s,1h),8.11(d,j=8.2hz,1h),7.77
–
7.69(m,2h),7.68
–
7.61(m,1h),7.48(d,j=5.5hz,1h),6.34(d,j=8.2hz,1h),3.01(t,j=7.2hz,2h),2.13(t,j=7.4hz,2h),1.68(dd,j=14.9,7.5hz,2h),1.59(dd,j=14.9,7.4hz,2h),1.45(dt,j=14.5,7.4hz,2h)。
[0166]
实施例13(制备12-3)
[0167]
结构式为化合物的制备方法同2-1。
[0168]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)δ8.52(s,1h),8.36(dd,j=11.3,6.9hz,2h),8.28(s,1h),8.11(d,j=8.3hz,1h),7.73(dd,j=14.4,7.7hz,2h),7.67
–
7.62(m,1h),7.49(d,j=5.4hz,1h),6.33(d,j=8.2hz,1h),3.03(s,2h),2.18(s,2h),1.71(s,4h)。
[0169]
实施例14(制备12-4)
[0170]
结构式为化合物的制备方法同2-1。
[0171]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)δ8.36(dd,j=12.8,7.0hz,2h),8.27(s,1h),8.10(d,j=8.2hz,1h),7.77
–
7.69(m,2h),7.68
–
7.61(m,1h),7.58(d,j=5.5hz,1h),6.34(d,j=8.2hz,1h),3.09
–
2.99(m,2h),2.27(t,j=7.2hz,2h),1.93(dt,j=12.0,5.8hz,2h)。
[0172]
实施例15(制备12-5)
[0173]
结构式为化合物的制备方法同2-1。
[0174]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)δ8.34(d,j=8.5hz,1h),8.30
–
8.25(m,2h),8.10(d,j=8.3hz,1h),7.97(s,1h),7.81(d,j=7.7hz,1h),7.75
–
7.69(m,2h),7.66
–
7.60(m,1h),7.52(d,j=5.4hz,1h),7.41(d,j=7.7hz,1h),7.28(t,j=7.6hz,1h),6.37(d,j=8.1hz,1h),4.30(s,2h)。
[0175]
实施例16(制备12-6)
[0176]
结构式为化合物的制备方法同2-1。
[0177]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)
δ8.34(d,j=8.5hz,1h),8.29
–
8.23(m,2h),8.09(d,j=8.2hz,1h),7.71(dd,j=7.7,5.4hz,2h),7.64
–
7.58(m,2h),7.56(dd,j=5.8,3.3hz,1h),7.40
–
7.34(m,1h),7.25
–
7.17(m,2h),6.39(d,j=8.1hz,1h),4.62(s,2h)。
[0178]
实施例17(制备12-7)
[0179]
结构式为化合物的制备方法同2-1。将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)δ8.35(d,j=8.5hz,1h),8.32
–
8.26(m,2h),8.13(d,j=8.2hz,1h),7.88(d,j=8.0hz,2h),7.74(dd,j=7.9,3.9hz,2h),7.68
–
7.63(m,1h),7.54(d,j=5.5hz,1h),7.38(d,j=8.0hz,2h),6.35(d,j=8.1hz,1h),4.31(s,2h)。
[0180]
实施例18(制备12-8)
[0181]
结构式为化合物的制备方法同2-1。
[0182]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)δ8.28(dd,j=10.3,5.2hz,3h),8.09(d,j=8.3hz,1h),7.71(t,j=7.4hz,2h),7.62(t,j=7.7hz,1h),7.55(d,j=6.2hz,2h),7.45(d,j=7.6hz,1h),7.25(d,j=7.8hz,1h),6.34(d,j=8.1hz,1h),4.24(s,2h),3.82(s,3h)。
[0183]
实施例19(制备12-9)
[0184]
结构式为化合物的制备方法同2-1。
[0185]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,d2o)δ8.03(d,j=4.8hz,1h),7.81(s,1h),7.55(d,j=8.2hz,1h),7.48(dd,j=11.6,4.9hz,1h),7.40(d,j=7.4hz,1h),7.28(s,1h),7.18(d,j=7.3hz,1h),7.15
–
7.12(m,1h),6.99(d,j=7.5hz,2h),6.95
–
6.85(m,5h),6.54(d,j=7.2hz,1h),5.73(d,j=7.7hz,1h),3.79(s,2h)。
[0186]
实施例20(制备13-1)
[0187]
结构式为化合物的制备方法如下:
[0188][0189]
在6-7(0.5mmol)的甲醇(5ml)溶液中加入由naoh(0.5mmol)水溶液0.5ml,室温下搅拌得到澄清溶液(通常过夜)。过滤掉不溶物,滤液旋蒸除去溶剂,残渣加入二氯甲烷(10ml x 3)共沸除去残余甲醇,再用ch2cl2洗涤,干燥后得到产物13-1。将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)δ8.11(dd,j=11.9,6.9hz,2h),7.98(d,j=8.0hz,1h),7.93(d,j=8.1hz,2h),7.70(dd,j=14.5,5.9hz,2h),7.60(t,j=7.4hz,1h),7.53
–
7.44(m,3h),7.38(d,j=8.0hz,1h),7.35
–
7.30(m,1h),4.44(s,2h),3.36(s,3h)。
[0190]
实施例21(制备13-2)
[0191]
结构式为化合物的制备方法同13-1。
[0192]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)δ8.55(s,2h),8.14(d,j=5.4hz,1h),8.08(d,j=8.3hz,1h),7.97(d,j=8.0hz,1h),7.69
–
7.64(m,2h),7.62(d,j=6.2hz,1h),7.59
–
7.52(m,3h),7.39(dd,j=13.1,8.0hz,2h),7.32
–
7.27(m,1h),4.40(s,2h),3.93(d,j=4.0hz,3h),3.34(s,3h)。
[0193]
实施例22(制备14-1)
[0194]
结构式为化合物的制备方法同13-1。将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)δ8.55(s,3h),8.35(d,j=8.5hz,1h),8.28(d,j=5.5hz,2h),8.09(d,j=8.4hz,1h),7.97(s,1h),7.81(d,j=7.5hz,
1h),7.75
–
7.68(m,2h),7.65
–
7.60(m,1h),7.52(d,j=5.4hz,1h),7.41(d,j=7.5hz,1h),7.28(t,j=7.6hz,1h),6.34(d,j=8.2hz,1h),4.29(s,2h)。
[0195]
实施例23(制备14-2)
[0196]
结构式为化合物的制备方法同13-1。
[0197]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,meod)δ8.55(s,2h),8.34(d,j=8.5hz,1h),8.30
–
8.22(m,2h),8.09(d,j=8.3hz,1h),7.71(dd,j=7.7,4.3hz,2h),7.61(dd,j=12.5,6.5hz,2h),7.57
–
7.53(m,1h),7.41
–
7.35(m,1h),7.24
–
7.18(m,2h),6.39(d,j=8.2hz,1h),4.63(s,2h)。
[0198]
实施例24(制备20-1)
[0199]
本实施例制备了x为2-亚甲基苯基时的吡啶3-羟基衍生物。式20-1的结构为:
[0200][0201]
制备方法由以下步骤组成:
[0202]
步骤一:化合物16的制备
[0203][0204]
将4-溴-1-萘甲腈(0.5g,2.16mmol),3-羟基-4-氯吡啶(0.28g,2.16mmol)加入dmf(15ml)中,加入cs2co3(2.11g,6.49mmol)和cui(催化量)。然后在80℃,氮气保护下搅拌反应混合物。直到tlc分析显示反应完全(一般在6小时内)。冷却至室温后,将反应物混合物倒入10ml冰水中,萃取至20x 3的乙酸乙酯中,用饱和食盐水洗涤有机层。用硅胶柱层析法对粗品进行纯化。所得白色固体化合物的收率为35%。1h nmr(300mhz,dmso-d6):δ8.71(s,1h),8.57(d,j=3.00hz,1h),8.49(d,j=6.00hz,1h),8.08(d,j=6.00hz,1h),7.94(t,j=6.00hz,1h),7.86(d,j=6.00hz,2h),6.80(d,j=6.00hz,1h)。
[0205]
步骤二:化合物的制备方法同5。
[0206]
步骤三:化合物的制备方法同6-1。
[0207]
步骤四:化合物的制备方法同7-1。
[0208]
步骤五:化合物的制备方法同8-1。
[0209]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,dmso)δ8.23(s,3h),7.92(d,j=6.6hz,1h),7.88(s,1h),7.77(s,2h),7.58(s,1h),7.44(s,1h),7.25(s,2h),7.17(s,1h),6.19(s,1h),5.77(s,2h)。
[0210]
实施例25(制备20-2)
[0211]
结构式为的制备方法同8-1。
[0212]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,dmso)δ8.23(s,2h),8.16(s,1h),7.96(d,j=7.0hz,1h),7.86(s,3h),7.66(d,j=18.6hz,1h),7.42(s,1h),7.30(s,3h),6.20(d,j=28.6hz,1h),5.29(d,j=13.6hz,2h)。
[0213]
实施例26(制备20-3)
[0214]
结构式为的制备方法同8-1。
[0215]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,dmso)δ8.23(s,3h),7.95(d,j=7.2hz,1h),7.91
–
7.85(m,1h),7.81
–
7.75(m,1h),7.71(d,j=8.0hz,1h),7.60(s,1h),7.47(s,2h),7.40(s,1h),6.20(d,j=5.6hz,1h),5.76(s,2h)。
[0216]
实施例27(制备20-4)
[0217]
结构式为的制备方法同8-1。
[0218]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,dmso)δ8.25(d,j=6.2hz,3h),8.02
–
7.94(m,3h),7.93
–
7.86(m,1h),7.84
–
7.76(m,1h),7.66(d,j=7.3hz,1h),7.39(s,1h),7.22(s,1h),6.26(d,j=4.9hz,1h),5.88(s,2h)。
[0219]
实施例28(制备20-5)
[0220]
结构式为的制备方法同8-1。
[0221]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,dmso)δ8.24(d,j=7.6hz,2h),7.99
–
7.92(m,1h),7.92
–
7.85(m,1h),7.80(d,j=7.4hz,1h),7.71
–
7.61(m,1h),7.51(s,1h),7.43(d,j=6.3hz,1h),7.31(s,1h),7.17(d,j=7.0hz,2h),6.20(d,j=5.7hz,1h),5.22(d,j=14.0hz,2h),3.78(s,3h)。
[0222]
实施例29(制备20-6)
[0223]
结构式为的制备方法同8-1。
[0224]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,dmso)δ8.23(dd,j=11.7,7.2hz,2h),8.14(d,j=7.5hz,1h),7.95
–
7.86(m,2h),7.81(d,j=7.7hz,1h),7.79
–
7.73(m,1h),7.67(dd,j=14.6,7.4hz,1h),7.60(d,j=11.4hz,1h),7.45(s,1h),6.84(d,j=8.2hz,1h),6.74(s,1h),5.81(d,j=15.4hz,2h),3.71(s,3h)。
[0225]
实施例30(制备20-7)
[0226]
结构式为的制备方法同8-1。
[0227]
将所得到的化合物采用核磁共振谱进行鉴定,鉴定结果为:1h nmr(400mhz,dmso)δ8.37(d,j=7.7hz,1h),8.25(t,j=7.5hz,1h),8.15(d,j=8.5hz,1h),7.93(dd,j=18.5,11.2hz,1h),7.82
–
7.74(m,1h),7.70(d,j=7.3hz,1h),7.67
–
7.61(m,1h),7.50
–
7.40(m,4h),7.39
–
7.33(m,3h),7.28(s,2h),7.21(d,j=6.4hz,1h),5.29(d,j=14.3hz,2h)。
[0228]
吡啶-3-胺衍生物对urat1抑制活性研究
[0229]
本发明化合物的urat1抑制效果采用如下方法测试所证明。
[0230]
这些效果表明本发明化合物对urat1抑制效果明显,其可用于治疗高尿酸血症。具体测试方法如下:
[0231]
一、实验目的及原理
[0232]
实验目的:通过比较加药前后hek293细胞对尿酸的重吸收作用来检测吡啶-3-胺衍生物对urat-1的抑制活性。
[0233]
实验原理:[14c]尿酸吸收缓冲液可用来模拟肾脏hurat1蛋白转运尿酸体内的离子交换环境。[14c]尿酸是用放射性[14c]同位素标记尿酸8位碳的尿酸分子。待细胞吸收[14c]尿酸后,通过测定细胞[14c]尿酸的放射值,计算药物对urat1抑制活性。
[0234]
二、试剂基本信息
[0235]
试剂名称品牌na-glumacklink-glumacklinca-glumacklin
glucosemacklinmgso4macklinkh2po4macklin
[0236]
三、试剂配制
[0237]
1.尿酸吸收缓冲液的配制
[0238]
用500ml超纯水按表2-1加入化合物配制[14c]尿酸吸收缓冲液,待所有成分经超声溶解后,用0.1m的naoh溶液和0.1m的hcl溶液调节ph至7.4,再用双圈定性滤纸抽滤除去杂质,于-20℃可保存一周。溶于500ml超纯水,ph=7.4。
[0239][0240]
2.药物配制
[0241]
a.[14c]尿酸吸收实验:以99.9%的dmso为溶剂,称取一定量的化合物和reda31370均溶解为50mm的母液,保存于-20℃,使用前用[14c]尿酸吸收缓冲液稀释成目的浓度。含药物缓冲液应与含[14c]尿酸的缓冲液以1:1混合达到实验最终所需吸收体积。药物终浓度的dmso含量不得高于0.2%。[14c]尿酸是用放射性[14c]同位素标记尿酸8位碳的尿酸分子。保存于-20℃,使用前用[14c]尿酸吸收缓冲液稀释成目的浓度。
[0242]
四、实验过程
[0243]
(1)用多聚赖氨酸包被24孔板,以增强hek293细胞的贴壁性;
[0244]
(2)将hek293稳转细胞株以15万个cells/500μl浓度接种到pdl包被的24孔板,用不含g418的完全培养基培养48h后,吸弃培养液,每孔加入500μl尿酸吸收缓冲液洗2次,再向每孔加入500μl尿酸吸收缓冲液孵育10min,随后迅速吸去孔内液体。加入含有不同浓度药物且含25μm的[14c]尿酸的吸收缓冲液开始吸收。
[0245]
(3)待[14c]尿酸吸收至20min时,吸弃孔内液体,加入500μl冰冷的dpbs;吸弃孔内液体,再加入700μl冰冷的dpbs;再吸弃孔内液体,加入1ml冰冷的dpbs。吸弃孔内液体,向孔内加入250μl 0.1mol/l的naoh溶液,室温下裂解20min。
[0246]
(4)向每个孔内中加入500μl闪烁液,用震荡仪震荡20min,混匀后用液体闪烁计数仪测定[14c]尿酸的放射值(cpm)。测定三次,取平均值。
[0247]
本发明中化合物的urat1的抑制活性结果如下表(以verinurad为对照组)。
[0248]
化合物8与13 urat1抑制活性
[0249][0250]
化合物12与14 urat1抑制活性
[0251][0252]
化合物20 urat1抑制活性
[0253][0254][0255]
对高尿酸血症小鼠(氧嗪酸钾与次黄嘌呤造模)治疗作用的体内药效学实验
[0256]
造模原理:氧嗪酸钾为尿酸酶抑制剂,可以抑制尿酸分解,次黄嘌呤在小鼠体内可代谢为尿酸,两者合用可以引起小鼠体内尿酸升高。
[0257]
实验方法:实验动物为体重20
±
2g的雄性昆明小鼠,设置空白对照组,模型组、给
药组和阳性药对照组,模型组12只,其余每组8只小鼠。小鼠适应性喂养七天后开始试验,实验前一天小鼠禁食处理,除空白对照组外,各组小鼠皮下注射400mg/kg氧嗪酸钾溶液(溶剂为0.5%cmc-na),空白对照组给予同等体积的0.5%cmc-na。0.5小时后给予空白对照组与模型组灌胃等量溶剂,各给药组则灌胃相应的药物与600mg/kg的次黄嘌呤。其中实施例9、16(化合物8-9,12-6)以10mg/kg剂量灌胃,阳性对照verinurad剂量也为10mg/kg。各组小鼠灌胃给予药物3h后,进行眼眶采血,血液静置30min后离心吸取血清,用尿酸试剂盒检测血尿酸水平,于-20℃保存。并同时将小鼠放入代谢笼,连续收集12小时内的尿液,测定尿液尿酸水平,于-80℃保存。
[0258]
实验结果:结果如图1和图2所示。
[0259]
图1中:***表示p《0.001与空白组比较,##表示p《0.01,###表示p《0.001与模型组比较。
[0260]
图2中:***表示p《0.001与空白组比较,##表示p《0.01,###表示p《0.001与模型组比较。
[0261]
图1所示为,以verinurad为阳性对照药,观察血尿酸水平。与模型相比,给与8-9,12-6治疗后的血尿酸水平明显降低,其中化合物12-6的降尿酸效果强于化合物8-9,且与阳性药verinurad效果相当。图2所示为,以verinurad为阳性对照药,给药后观察尿尿酸水平,与模型相比给予10mg/kg剂量的8-9,12-6后,尿尿酸水平明显增加,且与等剂量的verinurad相比,促尿酸排泄效果相当。
[0262]
上面结合实施例对本发明作了详细说明,但是本发明不限于上述实施例,在所属技术领域普通技术人员所具备的知识范围内,还可以在不脱离本发明宗旨的前提下作出各种变化。