1.本发明属于药物合成技术领域,具体涉及一种普克鲁胺中间体及其合成方法及由该中间体合成普克鲁胺的方法。
背景技术:
2.普克鲁胺(proxalutamide,gt-0918)是由苏州开拓药业研发的新一代ar(雌激素受体)拮抗剂,它是基于抗前列腺癌新药恩杂鲁胺(enzalutamide,mdv3100)的核心结构、采用基于靶蛋白晶体结构的计算机辅助设计并反复优化得到的新型化合物,该药的化学结构与恩杂鲁胺(mdv3100)相比较有了多处改变,从而改善了分子溶解度和动力学性质。新冠爆发以来,开拓药业将普克鲁胺用于covid-19的潜在治疗,结果显示可以显著降低新冠患者自轻症至重症的转化,2021年4月美国fda批准普克鲁胺开展治疗轻中症新冠男性患者的iii期临床试验,2021年7月巴拉圭国家公共卫生和社会福利部(mspbs)正式授予普克鲁胺紧急使用授权(eua),用于新冠住院患者的治疗;从现有临床数据来看,普克鲁胺治疗新冠患者的效果优异且安全性良好。
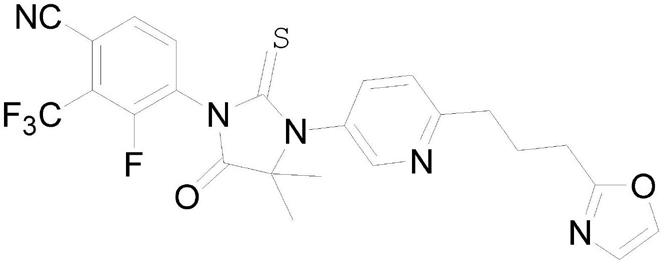
3.普克鲁胺的化学名是4-[4,4-二甲基-3-[6-[3-(2-恶唑基)丙基]-3-吡啶基]-5-羰基-2-硫代-1咪唑烷基]-3-氟-2-(三氟甲基)苯甲腈,cas号是1398046-21-3,化学结构如下:
[0004][0005]
目前公开的普克鲁胺的合成方法仅有苏州开拓药业的专利cn102482230和cn103608333,合成路线如下:
2-(三氟甲基)苯甲腈(化合物9);
[0025]
d、4-[2-[6-[3-(2-恶唑基)丙基]-3-吡啶基]胺基异丁酰胺基]-3-氟-2-(三氟甲基)苯甲腈(化合物9)与n,n'-硫羰基二咪唑(tcdi)反应,制得普克鲁胺。
[0026]
合成路线如下:
[0027][0028]
进一步地,所述步骤a中4-氨基-3-氟-2-(三氟甲基)苯甲腈、n-叔丁氧羰基-2-甲基丙氨酸、羰基二咪唑(cdi)和有机碱的摩尔比为1.0:(1.0-2.0):(1.0-2.0):(1.0-2.0),优选为1.0:(1.0-1.3):(1.0-1.2):(1.0-1.3),有机碱为三乙胺、二异丙基乙胺、吡啶、1,8-二氮杂二环十一碳-7-烯中(dbu)的一种,优选为1,8-二氮杂二环十一碳-7-烯(dbu)。
[0029]
进一步地,所述步骤b中4-(n-叔丁氧羰基-2-氨基异丁酰胺基)-3-氟-2-(三氟甲基)苯甲腈与氯化氢的异丙醇溶液的摩尔比为1.0:(5.0-20),优选为1.0:(5.0-1.0)。
[0030]
进一步地,所述步骤c中有机膦配体为p(o-tolyl)3、p(t-bu)3、binap、xantphos、brettphos、xphos等中的一种,优选为p(t-bu)3;无机碱为碳酸钾、碳酸铯、碳酸钠、叔丁醇钠等中的一种,优选为碳酸钾;4-(2-氨基异丁酰胺基)-3-氟-2-(三氟甲基)苯甲腈盐酸盐、2-[3-(2-恶唑基)丙基]-5-溴吡啶、醋酸钯、有机膦配体和无机碱的摩尔比为1.0:(0.8-1.2):(0.01-0.04):(0.01-0.3):(1.0-5.0),优选为1.0:(0.9-1.1):(0.015-0.035):(0.05-0.2):(1.5-3.5)。
[0031]
进一步地,所述步骤d中4-[2-[6-[3-(2-恶唑基)丙基]-3-吡啶基]胺基异丁酰胺基]-3-氟-2-(三氟甲基)苯甲腈与n,n'-硫羰基二咪唑的摩尔比为1.0:(1.0-1.5),优选为1.0:(1.0-1.2);有机溶剂为二氯甲烷、四氢呋喃、n,n-二甲基甲酰胺、甲苯、乙腈中的一种,优选为乙腈;4-[2-[6-[3-(2-恶唑基)丙基]-3吡啶基]胺基异丁酰胺基]-3-氟-2-(三氟甲基)苯甲腈在有机溶剂中浓度为0.05-0.20mol/l,优选为0.07-0.15mol/l。
[0032]
本发明具有以下有益效果:本发明首先以5-溴吡啶-2-乙酸为起始原料,经与米氏酸的酰化反应、羰基还原、亲核氨解脱羧和与碳酸亚乙烯酯的环合反应制备了普克鲁胺中间体2-[3-(2-恶唑基)丙基]-5-溴吡啶;然后以4-氨基-3-氟-2-(三氟甲基)苯甲腈和n-叔丁氧羰基-2-甲基丙氨酸为原料,经酰胺缩合反应、脱保护、buchwald-hartwig偶联反应和分子内的硫脲形成反应合成了普克鲁胺。相对于现有技术,本发明方法的合成步骤少、操作
简洁,避免使用剧毒的氰化物,降低了环境污染风险,本发明提供了合成普克鲁胺的新中间体2-[3-(2-恶唑基)丙基]-5-溴吡啶的制备方法,并以此中间体采用新路线合成了普克鲁胺,推进了原料药的工业化生产进程。
具体实施方式
[0033]
以下是本发明的具体实施例,对本发明的技术方案做进一步描述,但是本发明的保护范围并不限于这些实施例。凡是不背离本发明构思的改变或等同替代均包括在本发明的保护范围之内。
[0034]
实施例1、普克鲁胺中间体2-[3-(2-恶唑基)丙基]-5-溴吡啶(化合物1)的合成
[0035]
5-溴吡啶-2-乙酸(215g,1.0mol)、米氏酸(151.2g,1.05mol)和4-二甲氨基吡啶(dmap,183.25g,1.5mol)溶解到2.0l二氯甲烷中,降温至0℃,在1小时内慢慢分批加入二环己基碳二亚胺(dcc,226.8g,1.1mol),加完后继续保持0℃搅拌反应24小时。反应完成后,过滤除去沉淀,滤液依次用5%硫酸氢钾水溶液(1.0l*2)和饱和食盐水(500ml)洗涤,无水硫酸干燥16小时;过滤,向滤液中加入冰醋酸(600g,10.0mol),降温至0℃,慢慢分批加入硼氢化钠(94.5g,2.5mol),加完后继续在0℃搅拌反应24小时。反应完成后,将反应液倒入2l纯化水中,分液,有机层减压浓缩,残留物中加入1.5l乙酸乙酯溶解,依次用5%碳酸钠水溶液(500ml)、5%硫酸氢钾水溶液(500ml)和饱和食盐水(500ml)洗涤,无水硫酸钠干燥16小时,过滤,滤液减压浓缩,残留物用异丙醇/正庚烷(v/v,2/1)重结晶、真空干燥得到246.1克淡黄色固体5-(2-(5-溴吡啶-2-取代)乙基)-2,2-二甲基-1,3-二氧六环-4,6-二酮,收率75%。
[0036]
5l高压釜中,5-(2-(5-溴吡啶-2-取代)乙基)-2,2-二甲基-1,3-二氧六环-4,6-二酮(230g,0.7mol)加入到2.3l浓氨水(25%~28%)中,加热至100℃搅拌反应24小时;降温至5-10度,搅拌析晶2小时,抽滤,干燥,得到白色固体4-(5-溴吡啶-2-取代)丁酰胺160.5克,收率94.3%。
[0037]
氮气保护下,将4-(5-溴吡啶-2-取代)丁酰胺(158g,0.65mol)加入到500ml甲磺酸中,然后加入碳酸亚乙烯酯(86g,1.0mol),慢慢加热至120℃搅拌反应6小时。反应完成后直接减压浓缩,冷却到室温,残留物倒入1l纯化水中,冰浴冷却下用10%naoh调水溶液ph至10,继续搅拌1小时,抽滤得到黄色固体粗品,用异丙醚重结晶得到130.5克淡黄色固体2-[3-(2-恶唑基)丙基]-5-溴吡啶,收率75.2%。
[0038]
实施例2、普克鲁胺中间体2-[3-(2-恶唑基)丙基]-5-溴吡啶(化合物1)的合成
[0039]
6-溴吡啶-2-乙酸(215g,1.0mol)、米氏酸(144.1g,1.0mol)和4-二甲氨基吡啶(dmap,122.2g,1.0mol)溶解到2.0l二氯甲烷中,降温至0℃,在1小时内慢慢分批加入二环己基碳二亚胺(dcc,206.3g,1.0mol),加完后继续保持10℃搅拌反应18小时。反应完成后,过滤除去沉淀,滤液依次用5%硫酸氢钾水溶液(1.0l*2)和饱和食盐水(500ml)洗涤,无水硫酸干燥16小时;过滤,向滤液中加入冰醋酸(120g,2.0mol),降温至0℃,慢慢分批加入硼氢化钠(37.8g,1.0mol),加完后继续在10℃搅拌反应48小时。反应完成后,将反应液倒入2l纯化水中,分液,有机层减压浓缩,残留物中加入1.5l乙酸乙酯溶解,依次用5%碳酸钠水溶液(500ml)、5%硫酸氢钾水溶液(500ml)和饱和食盐水(500ml)洗涤,无水硫酸钠干燥16小时,过滤,滤液减压浓缩,残留物用异丙醇/正庚烷(v/v,2/1)重结晶、真空干燥得到229.7克
淡黄色固体5-(2-(5-溴吡啶-2-取代)乙基)-2,2-二甲基-1,3-二氧六环-4,6-二酮,收率70%。
[0040]
5l高压釜中,5-(2-(5-溴吡啶-2-取代)乙基)-2,2-二甲基-1,3-二氧六环-4,6-二酮(229.7g,0.7mol)加入到1.15l浓氨水(25%~28%)中,加热至120℃搅拌反应16小时;降温至5-10度,搅拌析晶2小时,抽滤,干燥,得到白色固体4-(5-溴吡啶-2-取代)丁酰胺152.7克,收率89.7%。
[0041]
氮气保护下,将4-(5-溴吡啶-2-取代)丁酰胺(150g,0.62mol)加入到300g甲磺酸中,然后加入碳酸亚乙烯酯(53.3g,1.0mol),慢慢加热至150℃搅拌反应2小时。反应完成后直接减压浓缩,冷却到室温,残留物倒入1l纯化水中,冰浴冷却下用10%naoh调水溶液ph至10,继续搅拌1小时,抽滤得到黄色固体粗品,用异丙醚重结晶得到121.5克淡黄色固体2-[3-(2-恶唑基)丙基]-5-溴吡啶,收率70.0%。
[0042]
实施例3、普克鲁胺中间体2-[3-(2-恶唑基)丙基]-5-溴吡啶(化合物1)的合成
[0043]
5-溴吡啶-2-乙酸(215g,1.0mol)、米氏酸(216.2g,1.5mol)和4-二甲氨基吡啶(dmap,244.4g,2.0mol)溶解到2.0l二氯甲烷中,降温至0℃,在1小时内慢慢分批加入二环己基碳二亚胺(dcc,309.5g,1.5mol),加完后继续保持-10℃搅拌反应48小时。反应完成后,过滤除去沉淀,滤液依次用5%硫酸氢钾水溶液(1.0l*2)和饱和食盐水(500ml)洗涤,无水硫酸干燥16小时;过滤,向滤液中加入冰醋酸(900g,15.0mol),降温至0℃,慢慢分批加入硼氢化钠(189g,5.0mol),加完后继续在-10℃搅拌反应20小时。反应完成后,将反应液倒入2l纯化水中,分液,有机层减压浓缩,残留物中加入1.5l乙酸乙酯溶解,依次用5%碳酸钠水溶液(500ml)、5%硫酸氢钾水溶液(500ml)和饱和食盐水(500ml)洗涤,无水硫酸钠干燥16小时,过滤,滤液减压浓缩,残留物用异丙醇/正庚烷(v/v,2/1)重结晶、真空干燥得到234.6克淡黄色固体5-(2-(5-溴吡啶-2-取代)乙基)-2,2-二甲基-1,3-二氧六环-4,6-二酮,收率71.5%。
[0044]
5l高压釜中,5-(2-(5-溴吡啶-2-取代)乙基)-2,2-二甲基-1,3-二氧六环-4,6-二酮(230g,0.7mol)加入到3.45l浓氨水(25%~28%)中,加热至80℃搅拌反应36小时;降温至5-10度,搅拌析晶2小时,抽滤,干燥,得到白色固体4-(5-溴吡啶-2-取代)丁酰胺156.7克,收率92.1%。
[0045]
氮气保护下,将4-(5-溴吡啶-2-取代)丁酰胺(150g,0.62mol)加入到750g甲磺酸中,然后加入碳酸亚乙烯酯(160g,1.86mol),慢慢加热至80℃搅拌反应12小时。反应完成后直接减压浓缩,冷却到室温,残留物倒入1l纯化水中,冰浴冷却下用10%naoh调水溶液ph至10,继续搅拌1小时,抽滤得到黄色固体粗品,用异丙醚重结晶得到118.9克淡黄色固体2-[3-(2-恶唑基)丙基]-5-溴吡啶,收率68.5%。
[0046]
实施例4、普克鲁胺的合成
[0047]
将羰基二咪唑(cdi,89.2g,0.55mol)慢慢加入到4-氨基-3-氟-2-(三氟甲基)苯甲腈(102.1g,0.5mol)和n-叔丁氧羰基-2-甲基丙氨酸(122g,0.6mol)的1.8l四氢呋喃溶液中,加热到60℃,滴加1,8-二氮杂二环十一碳-7-烯(dbu,91.3g,0.6mol),反应液继续保温搅拌反应4小时;减压浓缩,向残留物中加入1.5l二氯甲烷和20%柠檬酸水溶液,分液,有机相再用饱和食盐水洗涤一次,无水硫酸钠干燥,抽滤,滤液减压浓缩,得到180.6克白色固体4-(n-叔丁氧羰基-2-氨基异丁酰胺基)-3-氟-2-(三氟甲基)苯甲腈,收率92.8%。
[0048]
4-(n-叔丁氧羰基-2-氨基异丁酰胺基)-3-氟-2-(三氟甲基)苯甲腈(167.4g,0.43mol)加入到6n氯化氢的异丙醇溶液(500ml)中,室温搅拌反应24小时;过滤出不溶物,将滤液倒入2l异丙醚中,5度搅拌2小时,抽滤,得到124.1白色固体4-(2-氨基异丁酰胺基)-3-氟-2-(三氟甲基)苯甲腈盐酸盐,收率88.6%。
[0049]
5-(2-氨基异丁酰胺基)-3-氟-2-(三氟甲基)苯甲腈盐酸盐(114g,0.35mol)、2-[3-(2-恶唑基)丙基]-5-溴吡啶(93.5g,0.35mol)和碳酸钾(120.9g,0.875mol)悬浮在2.0l四氢呋喃中,搅拌15分钟,加入醋酸钯(1.96g,8.75mmol)和三叔丁基膦(p(t-bu)3,7.08g,35mmol),氮气保护下加热回流反应反应36小时;反应完成后冷却到室温,将反应液倒入1.0l饱和食盐水和2.0l乙酸乙酯的混合液中,分出有机层,无水硫酸钠干燥,抽滤,滤液减压浓缩,残留物用甲醇和水重结晶得到淡黄色固体4-[2-[6-[3-(2-恶唑基)丙基]-3-吡啶基]胺基异丁酰胺基]-3-氟-2-(三氟甲基)苯甲腈133.2克,收率80.05%。
[0050]
实验证明有机膦配体p(t-bu)3可以用p(o-tolyl)3、binap、xantphos、brettphos、xphos中的一种来代替;无机碱碳酸钾可以用碳酸铯、碳酸钠、叔丁醇钠中的一种来代替;4-(2-氨基异丁酰胺基)-3-氟-2-(三氟甲基)苯甲腈盐酸盐、2-[3-(2-恶唑基)丙基]-5-溴吡啶、醋酸钯、有机膦配体和无机碱的摩尔比可以是1.0:(0.8-1.2):(0.01-0.04):(0.01-0.3):(1.0-5.0)。
[0051]
4-[2-[6-[3-(2-恶唑基)丙基]-3-吡啶基]胺基异丁酰胺基]-3-氟-2-(三氟甲基)苯甲腈(95.1g,0.2mol)溶解到500ml乙腈中,加入n,n'-硫羰基二咪唑(tcdi,39.2g,0.22mol),氮气保护下回流反应20小时。反应液减压浓缩,加入1.0l二氯甲烷溶解,依次用饱和碳酸氢钠水溶液和饱和食盐水洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩,所得残留物用异丙醇和水重结晶得到79.6克淡黄色固体产品普克鲁胺,收率76.9%。
[0052]
实施例5、普克鲁胺的合成
[0053]
将羰基二咪唑(cdi,81.1g,0.5mol)慢慢加入到4-氨基-3-氟-2-(三氟甲基)苯甲腈(102.1g,0.5mol)和n-叔丁氧羰基-2-甲基丙氨酸(101.7g,0.5mol)的1.8l四氢呋喃溶液中,加热到60℃,滴加1,8-二氮杂二环十一碳-7-烯(dbu,76.1g,0.5mol),反应液继续保温搅拌反应4小时;减压浓缩,向残留物中加入1.5l二氯甲烷和20%柠檬酸水溶液,分液,有机相再用饱和食盐水洗涤一次,无水硫酸钠干燥,抽滤,滤液减压浓缩,得到175.5克白色固体4-(n-叔丁氧羰基-2-氨基异丁酰胺基)-3-氟-2-(三氟甲基)苯甲腈,收率90.2%。
[0054]
4-(n-叔丁氧羰基-2-氨基异丁酰胺基)-3-氟-2-(三氟甲基)苯甲腈(167.4g,0.43mol)加入到6n氯化氢的异丙醇溶液(360ml)中,室温搅拌反应24小时;过滤出不溶物,将滤液倒入2l异丙醚中,5度搅拌2小时,抽滤,得到121.5白色固体4-(2-氨基异丁酰胺基)-3-氟-2-(三氟甲基)苯甲腈盐酸盐,收率86.7%。
[0055]
(2-氨基异丁酰胺基)-3-氟-2-(三氟甲基)苯甲腈盐酸盐(114g,0.35mol)、2-[3-(2-恶唑基)丙基]-5-溴吡啶(74.8g,0.28mol)和碳酸钾(48.4g,0.35mol)悬浮在2.0l四氢呋喃中,搅拌15分钟,加入醋酸钯(0.78g,3.5mmol)和三叔丁基膦(p(t-bu)3,0.71g,3.5mmol),氮气保护下加热回流反应反应36小时;反应完成后冷却到室温,将反应液倒入1.0l饱和食盐水和2.0l乙酸乙酯的混合液中,分出有机层,无水硫酸钠干燥,抽滤,滤液减压浓缩,残留物用甲醇和水重结晶得到淡黄色固体4-[2-[6-[3-(2-恶唑基)丙基]-3-吡啶基]胺基异丁酰胺基]-3-氟-2-(三氟甲基)苯甲腈100.5克,收率60.7%。
[0056]
4-[2-[6-[3-(2-恶唑基)丙基]-3-吡啶基]胺基异丁酰胺基]-3-氟-2-(三氟甲基)苯甲腈(95.1g,0.2mol)溶解到400ml乙腈中,加入n,n'-硫羰基二咪唑(tcdi,35.6g,0.2mol),氮气保护下回流反应20小时。反应液减压浓缩,加入1.0l二氯甲烷溶解,依次用饱和碳酸氢钠水溶液和饱和食盐水洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩,所得残留物用异丙醇和水重结晶得到68.7克淡黄色固体产品普克鲁胺,收率66.4%。
[0057]
实施例6、普克鲁胺的合成
[0058]
将羰基二咪唑(cdi,162.2g,1.0mol)慢慢加入到4-氨基-3-氟-2-(三氟甲基)苯甲腈(102.1g,0.5mol)和n-叔丁氧羰基-2-甲基丙氨酸(203.3g,1.0mol)的1.8l四氢呋喃溶液中,加热到60℃,滴加1,8-二氮杂二环十一碳-7-烯(dbu,152.2g,1.0mol),反应液继续保温搅拌反应4小时;减压浓缩,向残留物中加入1.5l二氯甲烷和20%柠檬酸水溶液,分液,有机相再用饱和食盐水洗涤一次,无水硫酸钠干燥,抽滤,滤液减压浓缩,得到170.5克白色固体4-(n-叔丁氧羰基-2-氨基异丁酰胺基)-3-氟-2-(三氟甲基)苯甲腈,收率87.6%。
[0059]
4-(n-叔丁氧羰基-2-氨基异丁酰胺基)-3-氟-2-(三氟甲基)苯甲腈(167.4g,0.43mol)加入到6n氯化氢的异丙醇溶液(1400ml)中,室温搅拌反应24小时;过滤出不溶物,将滤液倒入14l异丙醚中,5度搅拌2小时,抽滤,得到122.5白色固体4-(2-氨基异丁酰胺基)-3-氟-2-(三氟甲基)苯甲腈盐酸盐,收率87.5%。
[0060]
6-(2-氨基异丁酰胺基)-3-氟-2-(三氟甲基)苯甲腈盐酸盐(114g,0.35mol)、2-[3-(2-恶唑基)丙基]-5-溴吡啶(112.2g,0.42mol)和碳酸钾(241.8g,1.75mol)悬浮在2.0l四氢呋喃中,搅拌15分钟,加入醋酸钯(3.14g,14mmol)和三叔丁基膦(p(t-bu)3,21.2g,105mmol),氮气保护下加热回流反应反应36小时;反应完成后冷却到室温,将反应液倒入1.0l饱和食盐水和2.0l乙酸乙酯的混合液中,分出有机层,无水硫酸钠干燥,抽滤,滤液减压浓缩,残留物用甲醇和水重结晶得到淡黄色固体4-[2-[6-[3-(2-恶唑基)丙基]-3-吡啶基]胺基异丁酰胺基]-3-氟-2-(三氟甲基)苯甲腈113.5克,收率68.2%。
[0061]
4-[2-[6-[3-(2-恶唑基)丙基]-3-吡啶基]胺基异丁酰胺基]-3-氟-2-(三氟甲基)苯甲腈(95.1g,0.2mol)溶解到4.0l乙腈中,加入n,n'-硫羰基二咪唑(tcdi,53.5g,0.3mol),氮气保护下回流反应20小时。反应液减压浓缩,加入1.0l二氯甲烷溶解,依次用饱和碳酸氢钠水溶液和饱和食盐水洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩,所得残留物用异丙醇和水重结晶得到70.5克淡黄色固体产品普克鲁胺,收率68.1%。
[0062]
本发明不局限于上述实施方式,任何人应得知在本发明的启示下作出的结构变化,凡是与本发明具有相同或相近的技术方案,均落入本发明的保护范围之内。
[0063]
本发明未详细描述的技术、形状、构造部分均为公知技术。