1.本发明涉及药物相关杂质合成技术领域,尤其涉及一种头孢卡品酸的合成方法。
背景技术:
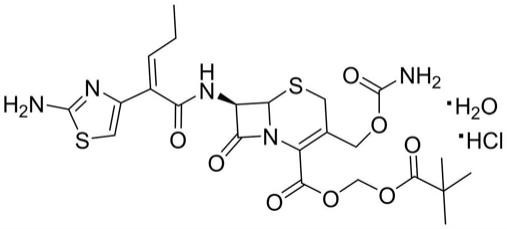
2.盐酸头孢卡品酯(cefcapene pivoxil hydrochloride hydrate)是第三代口服头孢抗生素,属广谱抗生素类药物,由日本盐野义制药株式会社开发,对革兰氏阳性菌和阴性菌均表现出广谱的抗菌作用,对可以水解抗生素的β-内酰胺酶高度稳定。
3.盐酸头孢卡品酯的化学名称为:7-[2-(2-氨基-1,3-噻唑-4-基)戊-3-烯酰胺基]-3-(氨基甲酰氧甲基)-8-氧代-5-硫-1-氮杂双环[4.2.0]辛-2-烯-2-甲酸2,2-二甲基丙酰氧甲基酯盐酸盐,其化学结构如下:
[0004][0005]
目前报道的合成盐酸头孢卡品酯的方法较多,但大多数工艺都与 w02008155615中报道的方法相似,以羟甲基-7-氨基头孢烷酸为起始物料,经与侧链缩合反应、3号位羟基胺甲酰化反应和水解反应后得到头孢卡品酯前体酸bcn,再经与碘酯酯化反应和脱除boc后,得到盐酸头孢卡品酯。在脱除boc过程中,极易产生杂质头孢卡品酸,且在加速实验中发现,头孢卡品酸降解出来的速度比较快,研究该杂质的控制因素对提高盐酸头孢卡品酯的质量具有重要意义。
[0006]
目前报道的关于头孢卡品酸的研究不多,现有报道的合成方法中,制备的头孢卡品酸中含有较多的杂质,纯度较低。
技术实现要素:
[0007]
有鉴于此,本发明提供了一种头孢卡品酸的合成方法。本发明提供的合成方法产物纯度高。
[0008]
为了实现上述发明目的,本发明提供以下技术方案:
[0009]
一种头孢卡品酸的合成方法,包括以下步骤:
[0010]
(1)将bcn、有机溶剂和无机酸混合进行酸化反应,将所得酸化料液静置分层,得到含有脱盐bcn的有机相;
[0011]
所述bcn为头孢卡品酯前体酸,结构式如式i所示;所述脱盐bcn的结构式如式ii所示:
[0012][0013]
(2)将所述含有脱盐bcn的有机相和酸混合进行叔丁氧羰基脱除反应,所得产物料液进行固液分离,得到头孢卡品酸粗产物;
[0014]
(3)将所述头孢卡品酸粗产物分散于水中,用碱将所得分散液的ph值调节至8.0~9.0,然后进行固液分离,得到滤液;
[0015]
(4)用酸将所述滤液的ph值调节至2~4,使晶体析出,然后进行降温养晶,得到头孢卡品酸。
[0016]
优选的,所述步骤(1)中的有机溶剂包括乙酸乙酯、二氯甲烷、甲醇、乙醇、n,n二甲基甲酰胺和n,n二甲基乙酰胺中的一种或几种;所述bcn 和有机溶剂的用量比为1g:5~10ml。
[0017]
优选的,所述步骤(1)中的无机酸包括硫酸、盐酸和磷酸中的一种或几种;所述硫酸、盐酸和磷酸的质量分数独立地为5~25%;所述酸化反应的 ph值为1.5~2,所述酸化反应的时间为20~40min。
[0018]
优选的,所述步骤(2)中酸为盐酸、三氟乙酸和四氯化钛中的一种或几种。
[0019]
优选的,所述步骤(2)中酸和步骤(1)中bcn的摩尔比为(15~30): 1。
[0020]
优选的,所述叔丁氧羰基脱除反应的温度为-5~5℃,时间为20~40min。
[0021]
优选的,所述步骤(3)中头孢卡品酸粗产物和水的用量比为1g: 10~20ml。
[0022]
优选的,所述步骤(3)中的碱为氢氧化钠、氢氧化钾、碳酸氢钠和碳酸钠中的一种或几种。
[0023]
优选的,所述步骤(4)中的酸为盐酸或硫酸。
[0024]
优选的,所述降温养晶的温度为0~5℃,时间为45~90min。
[0025]
本发明提供了一种头孢卡品酸的合成方法,本发明以bcn为原料,先用酸中和bcn中的二异丙胺盐得到脱盐bcn,然后脱除boc保护基,得到头孢卡品酸粗品,之后用碱纯化,去除头孢卡品酸与无机酸形成的盐和一些碱溶性杂质,最后在酸性条件下降温析晶,酸溶性杂质被留在母液中,析出的晶体为高纯度的头孢卡品酸。本发明提供的方法简便高效,容易操作,获得的高纯度头孢卡品酸可作为对照品对盐酸头孢卡品酯中该关键杂质进行分析,为盐酸头孢卡品酯的质量控制起到积极作用。
[0026]
进一步的,本发明采用盐酸、三氟乙酸或四氯化钛脱除脱盐bcn中的 boc基团,boc的脱除效率高,并且本发明整个合成过程中没有用到高浓度磷酸,环保压力小。现有技术中采用高浓度磷酸脱除boc基团,容易产生难以处理的磷酸废水,且boc脱除效率低。
[0027]
实施例结果表明,采用本发明的方法合成的头孢卡品酸纯度为98%以上。
附图说明
[0028]
图1为实施例1制备的头孢卡品酸的hplc色谱图。
具体实施方式
[0029]
本发明提供了一种头孢卡品酸的合成方法,包括以下步骤:
[0030]
(1)将bcn、有机溶剂和无机酸混合进行酸化反应,将所得酸化料液静置分层,得到含有脱盐bcn的有机相;所述bcn为头孢卡品酯前体酸,结构式如式i所示;所述脱盐bcn的结构式如式ii所示:
[0031][0032]
(2)将所述含有脱盐bcn的有机相和酸混合进行叔丁氧羰基脱除反应,所得产物料液进行固液分离,得到头孢卡品酸粗产物;
[0033]
(3)将所述头孢卡品酸粗产物分散于水中,用碱将所得分散液的ph值调节至8.0~9.0,然后进行固液分离,得到滤液;
[0034]
(4)用酸将所述滤液的ph值调节至2~4,使晶体析出,然后进行降温养晶,得到头孢卡品酸。
[0035]
本发明将bcn、有机溶剂和无机酸混合进行酸化反应,将所得酸化料液静置分层,得到含有脱盐bcn的有机相。在本发明中,所述bcn优选为 bcn粗品,所述bcn粗品的纯度优选为98.5%及以上;本发明对所述bcn 粗品的来源没有特殊要求,采用市售的bcn粗品或自行制备均可;所述有机溶剂包括乙酸乙酯、二氯甲烷、甲醇、乙醇、n,n二甲基甲酰胺和n,n二甲基乙酰胺中的一种或几种;所述bcn和有机溶剂的用量比为1g:5~10ml;所述无机酸优选包括硫酸、盐酸和磷酸中的一种或几种,所述硫酸、盐酸和磷酸的质量分数独立地优选为5~25%,更优选为10%;所述酸化反应的ph 值优选为1.5~2。在本发明中,所述酸化反应的温度优选为10~25℃,更优选为15~20℃;所述酸化反应的时间优选为20~40min,更优选为30min。
[0036]
在本发明的具体实施例中,优选在10~25℃条件下,将bcn粗品加入有机溶剂中,搅拌均匀,然后加入有机酸将料液的ph值调节至1.5~2,之后搅拌至体系溶清。在酸化反应过程中,bcn发生酸化中和,由二乙丙胺盐形式转变为base结构。
[0037]
本发明对所述静置分层没有特殊要求,采用本领域技术人员熟知的方法即可。静置分层后,所得有机层的主要成分为脱盐bcn和有机溶剂。
[0038]
得到含有脱盐bcn的有机相后,本发明将所述含有脱盐bcn的有机相和酸混合进行叔丁氧羰基脱除反应,所得产物料液进行固液分离,得到头孢卡品酸粗产物。在本发明中,所述酸优选为盐酸、三氟乙酸和四氯化钛中的一种或几种,其中四氯化钛为路易斯酸;所述盐酸优选为浓盐酸;所述步骤 (2)中的酸和步骤(1)中的bcn的摩尔比优选为(15~30):1。
[0039]
在本发明中,所述叔丁氧羰基脱除反应的温度优选为-5~5℃,反应时间优选为20~40min,更优选为30min。
[0040]
在本发明的具体实施例中,优选在-5~5℃条件下,将酸滴加到含有脱盐 bcn的有机相中,反应至有晶体析出,反应完成后,将所得产物料液进行固液分离,得到头孢卡品酸粗产物;所述固液分离的方式优选为过滤。
[0041]
得到头孢卡品酸粗产物后,本发明将所述头孢卡品酸粗产物分散于水中,用碱将所得分散液的ph值调节至8.0~9.0,然后进行固液分离,得到滤液。在本发明中,所述水优选为纯水;所述头孢卡品酸粗产物和水的用量比优选为1g:10~20ml;碱为氢氧化钠、氢氧化钾、碳酸氢钠和碳酸钠中的一种或几种;所述碱优选以碱溶液的形式使用,所述碱溶液的溶剂优选为水,所述碱溶液的质量浓度优选为7~10%。在本发明的具体实施例中,优选在 10~25℃条件下,将头孢卡品酸粗产物和水混合,搅拌均匀后,再滴加碱溶液,将料液的ph值调节至8.0~9.0,优选调节至8.3~8.5,然后继续搅拌30min,之后进行过滤,得到滤液。头孢卡品酸粗品中混有头孢卡品酸与无机酸形成的盐以及碱溶性杂质,本发明用碱纯化,可有效去除头孢卡品酸与无机酸形成的盐以及碱溶性杂质。
[0042]
得到滤液后,本发明用酸将所述滤液的ph值调节至2~4,使晶体析出,然后进行降温养晶,得到头孢卡品酸。在本发明中,所述酸优选为盐酸或硫酸;所述盐酸的质量浓度优选为3~4%,更优选为3.6%,所述硫酸的质量浓度优选为3~4%,更优选为3.6%。本发明用酸将所述滤液的ph值调节至2~4,优选调节至2.5~3.5,调节到上述ph值后,头孢卡品酸晶体从溶液中析出,而酸溶性杂质则留在溶液中,从而实现头孢卡品酸的进一步纯化。
[0043]
在本发明中,所述降温养晶的温度优选为0~5℃,时间优选为45~90min,更优选为60min。降温养晶完成后,本发明优选将料液过滤,将所得固体产物减压干燥,得到头孢卡品酸;所述减压干燥的温度优选为30℃,时间优选为3~5h,在本发明中,所述头孢卡品酸的结构式如式iii所示:
[0044][0045]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。
[0046]
实施例1
[0047]
在25℃下,向烧瓶中加入bcn粗品10g,二氯甲烷100ml,搅拌均匀后,滴加稀盐酸至ph值为1.5,搅拌至体系溶清,静置分层,向二氯甲烷层中再滴加浓盐酸20ml,有晶体明显析出,反应60min;过滤,使用20ml 纯水洗涤滤饼;在25℃下,向烧瓶中加入滤饼与100ml的纯水,搅拌均匀后,滴加7%碳酸氢钠溶液,调节ph值至8,搅拌30min,过滤,收集滤液;向滤液中滴加3.6%的盐酸,调节ph值至2,析出晶体,降温5℃,养晶60min,过滤,抽干后,转至烘箱30℃减压干燥3h;得到干品6.7g,产品纯度为98.4%。产物的hplc检测数据见表1,hplc色谱图见图1。
[0048]
表1为实施例1制得的头孢卡品酸的hplc检测数据 (检测器波长为265nm)
[0049]
峰号保留时间高度面积面积%理论塔板数usp拖尾因子13.19110392846311.57131141.40825.520392639530117098.42938601.259总计 4030315385800100.000
ꢀꢀ
[0050]
实施例2
[0051]
在25℃下,向烧瓶中加入bcn粗品10g,乙酸乙酯100ml,搅拌均匀后,滴加稀盐酸至
ph值为2.0,搅拌至体系溶清,静置分层,向乙酸乙酯层中再滴加浓盐酸20ml,有晶体明显析出,反应60min;过滤,使用20ml 纯水洗涤滤饼;在25℃下,向烧瓶中加入滤饼与100ml的纯水,搅拌均匀后,滴加7%碳酸氢钠溶液,调节ph值至9,搅拌30min,过滤,收集滤液;向滤液中滴加3.6%的盐酸,调节ph至4,析出晶体,降温至5℃,养晶60min,过滤,抽干后,转至烘箱30℃减压干燥5h;得到干品5.5g,产品纯度为98.3%。
[0052]
实施例3
[0053]
在20℃下,向烧瓶中加入bcn粗品10g,二氯甲烷100ml,搅拌均匀后,滴加稀盐酸至ph值为2.0,搅拌至体系溶清,静置分层,向二氯甲烷层中再滴加浓盐酸20ml,有晶体明显析出,反应60min;过滤,使用20ml 纯水洗涤滤饼;在20℃下,500ml烧瓶中加入滤饼与100ml的纯水,搅拌均匀后,滴加10%氢氧化钠溶液,调节ph值至10,搅拌30min,过滤,收集滤液;向滤液中滴加3.6%的硫酸,调节ph值至3,析出晶体,降温至3℃,养晶60min,过滤,抽干后,转至烘箱30℃减压干燥4h;得到干品5.9g,产品纯度为98.2%。
[0054]
实施例4
[0055]
在室温下,向烧瓶中加入bcn粗品10g,二氯甲烷100ml,搅拌均匀后,滴加稀盐酸至ph值为2.0,搅拌至体系溶清,静置分层,向二氯甲烷层中再滴加浓盐酸10ml,有晶体明显析出,反应60min;过滤,使用20ml 纯水洗涤滤饼;在室温下,500ml烧瓶中加入滤饼与100ml的纯水,搅拌均匀后,滴加10%氢氧化钠溶液,调节ph值至9,搅拌30min,过滤,收集滤液;向滤液中滴加3.6%的硫酸,调节ph至2.5,析出晶体,降温至5℃,养晶60min,过滤,抽干后,转至烘箱30℃减压干燥5h;得到干品6.5g,产品纯度为98.2%。
[0056]
实施例5
[0057]
在10℃下,向烧瓶中加入bcn粗品10g,乙酸乙酯100ml,搅拌均匀后,滴加稀盐酸至ph值至1.5,搅拌至体系溶清,静置分层,向乙酸乙酯层中再滴加浓盐酸10ml,有晶体明显析出,反应60min;过滤,使用20ml 纯水洗涤滤饼;在10℃下,向烧瓶中加入滤饼与100ml的纯水,搅拌均匀后,滴加10%氢氧化钠溶液,调节ph值至8,搅拌30min,过滤,收集滤液;向滤液中滴加3.6%的硫酸,调节ph至2,析出晶体,降温至0℃,养晶60min,过滤,抽干后,转至烘箱30℃减压干燥3h;得到干品6.6g,产品纯度为98.4%。
[0058]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。