1.本发明属于镜片用材料技术领域,具体的说,涉及一种吲哚类化合物及其在制备镜片中的应用。
背景技术:
2.2010年后,智能手机、平板、电脑等逐渐进入人们生活中,各种手机app等占用着人们大量的碎片化时间,而这一变化会对人们的眼睛产生巨大的负担。一般来说,电子设备的屏幕会持续散发有危害性的蓝光辐射,其中波长处于380~450nm之间的高能短波蓝光对视网膜存在永久性损伤风险。这类蓝光辐射具有很强的穿透力,能够穿透晶状体直达视网膜,引起视网膜黄斑部病变,加剧色差和视觉模糊,最终破坏激素分泌平衡并影响视力和睡眠质量,随之而来的各种问题和隐患也会越发的明显。因此,各种针对预防和治疗有害蓝光的技术在近年来取得了不少进展。特别是各种防蓝光屏、防蓝光膜和防蓝光眼镜技术,可通过减少智能设备对透射至人眼的蓝光来起到保护作用,减少危害。
3.常见的防蓝光技术是通过添加一种或几种蓝光吸收剂来达到降低蓝光透射率的作用,常用的蓝光吸收剂包括色粉黄色、色粉兰色、色粉桔红、纳米al2o3/聚丙烯酸混合物、纳米tio2/聚丙烯酸混合物、纳米fe2o3/聚丙烯酸混合物、金属络合类染料、苯并三唑类化合物、pmma聚甲基丙烯酸和富勒烯等。这些吸收剂中,像色粉存在着底色深、透光率降低、显著影响人们日常使用的缺陷;而纳米无机材料存在制作工艺复杂、制备成本高的问题;剩下其他几类吸收剂则存在防蓝光效果不明显的缺点。
4.因此,开发低成本、易于工业化生产并具备高效率高寿命的有机蓝光吸收剂,应用于防蓝光镜片和防蓝光膜具有明显的商业价值。
技术实现要素:
5.本发明的第一个目的是提供一种吲哚类化合物。
6.本发明的第二个目的是提供一种所述吲哚类化合物在制备蓝光吸收剂中的应用。
7.本发明的第三个目的是提供一种所述吲哚类化合物在制备防蓝光基片镜片中的应用。
8.为了实现上述目的,本发明采用的技术方案如下:
9.本发明的第一方面提供了一种吲哚类化合物,具有以下结构通式:
[0010][0011]
其中,r1选自氢、c1~c8的直链烷基、c1~c8含有支链的烷基、r3选自氢、甲基、乙基、甲氧基、乙氧基;
[0012]
r2选自氢、c1~c8的直链烷基、c1~c8含有支链的烷基。
[0013]
较优选的,所述吲哚类化合物中,r1选自氢、甲基、乙基、异丙基、正丙基、正丁基、叔丁基、r3选自氢、甲基、乙基、甲氧基、乙氧基;
[0014]
r2选自氢、甲基、乙基、异丙基、正丙基、正丁基、叔丁基。
[0015]
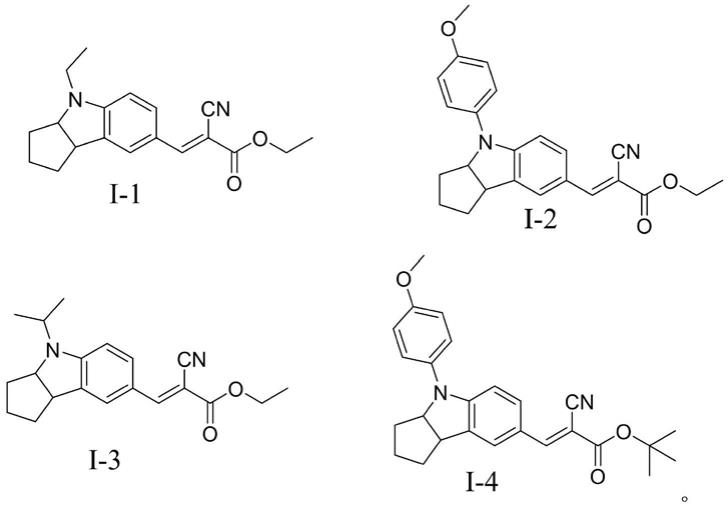
最优选的,所述吲哚类化合物的结构选自以下结构的一种:
[0016][0017][0018]
本发明的第二方面提供了一种所述吲哚类化合物在制备蓝光吸收剂中的应用。
[0019]
本发明的第三方面提供了一种防蓝光基片镜片,是由以下重量份的组分制成:单官能基反应单体25-75份、多官能基反应单体25-75份、低聚物0-10份,并加入引发剂和蓝光吸收剂,所述引发剂的加入量占单官能基反应单体、多官能基反应单体、低聚物、引发剂和蓝光吸收剂总质量的0.1%-0.15%,所述蓝光吸收剂加入量占单官能基反应单体、多官能基反应单体、低聚物、引发剂和蓝光吸收剂总质量的0.01%-0.15%。
[0020]
所述低聚物优选为1、2、3、4~10份。
[0021]
较优选的,所述防蓝光基片镜片是由以下重量份的组分制成:单官能基反应单体50-75份、多官能基反应单体25-45份、低聚物5-10份,并加入引发剂和蓝光吸收剂,所述引发剂的加入量占单官能基反应单体、多官能基反应单体、低聚物、引发剂和蓝光吸收剂总质量的0.1%,所述蓝光吸收剂加入量占单官能基反应单体、多官能基反应单体、低聚物、引发剂和蓝光吸收剂总质量的0.1%。
[0022]
所述单官能基反应单体选自苯乙烯、甲基丙烯酸、丙烯酸、甲基苯乙烯、丙烯酸丁酯、丙烯酸异辛酯、甲基丙烯酸乙酯、丙烯酸环己酯、苄基丙烯酸酯、乙氧化壬基苯酚丙烯酸酯、丙烯酸羟乙酯、邻苯基苯氧基乙基丙烯酸酯、四氢化糠基丙烯酸酯等。
[0023]
所述多官能基反应单体选自二丙二醇二丙烯酸酯、三丙二醇二丙烯酸酯、聚乙二醇二丙烯酸酯、乙氧化双酚a二丙烯酸酯、聚乙二醇二甲基丙烯酸酯、丙氧化甘油三丙烯酸酯、季戊四醇四丙烯酸酯、乙氧化季戊四醇四丙烯酸酯等。
[0024]
所述低聚物选自脂肪族聚氨酯丙烯酸酯低聚物、芳香族聚氨酯丙烯酸酯、环氧丙
烯酸酯、双酚a环氧丙烯酸酯、聚酯丙烯酸酯等。
[0025]
所述引发剂选自过氧化二碳酸二异丙酯、过氧化二碳酸二环己酯、过氧化苯甲酰、过氧化二异丙苯、过氧化苯甲酸叔丁酯、偶氮二异丁腈、偶氮二异庚腈。
[0026]
所述蓝光吸收剂为所述吲哚类化合物。
[0027]
所述防蓝光基片镜片的制备方法包括以下步骤:
[0028]
第一步,将单官能基反应单体、多官能基反应单体、低聚物、蓝光吸收剂混合均匀,加入引发剂,在室温下抽真空10~60min,再将温度控制在35~50℃,静置抽真空10~60min;
[0029]
第二步,把混合均匀的第一步制备的混合物过滤,注入模具中密封,在固化炉中经过20小时从室温程序升温至95℃,完成一次固化获得镜片;
[0030]
程序升温曲线:
①
0-5h保持34℃;
②
5-12h匀速升至50℃;
③
12-18h匀速升至95℃;
④
18-20h保持95℃;
[0031]
第三步,将第二步获得的镜片进行开模、切边和清洗工序,在温度为105~115℃的条件下恒温1~2小时进行二次固化成型,得到所述防蓝光基片镜片。
[0032]
由于采用上述技术方案,本发明具有以下优点和有益效果:
[0033]
本发明的防蓝光基片镜片采用本发明的吲哚类化合物,防蓝光效果显著,蓝光吸收剂添加量大大减少,基本不影响镜片的各项物理性能。
[0034]
本发明的防蓝光基片镜片,在相同测试条件下具有较低的黄色指数(这里需要指出的是,本发明的设备测的黄色指数的值都有点偏高,是内部测出来的值,所以进行的是内部的平行比较)。
[0035]
本发明的防蓝光基片镜片,具有耐黄变性,经长时间的光照,黄色指数不发生变化。
附图说明
[0036]
图1是化合物i-1和化合物i-2的thf溶液(1%质量分数)的吸光度示意图。
具体实施方式
[0037]
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
[0038]
下列实施例中所说的室温是指(20℃~30℃)。
[0039]
本发明所使用的有机溶剂如无水四氢呋喃(thf)、n,n-二甲基甲酰胺(dmf)和乙醇等均采购自阿拉丁,1,2,3,3a,4,8b-六氢环戊[b]吲哚购自北京百灵威科技有限公司,其他反应试剂购自sigma-aldrich。
[0040]
本发明的黄色指数和截止波长测试仪器均为安捷伦agilent cary 60型紫外-可见分光光谱仪,落球抗冲击性能为labthink的bmc-b1型落球冲击试验机,抗老化性能测试仪器为q-lab的q-sun xe-1型xenon test chamber。
[0041]
实施例1
[0042][0043]
第一步,将干燥的式(ii)所示化合物(15.9g,0.1mol)与溴乙烷(10.9g,0.1mol)溶于230ml无水四氢呋喃中,加入干燥的四口烧瓶中,室温搅拌2h,将反应液倾倒入大量水中,二氯甲烷萃取(100ml),水洗五次(5
×
100ml),有机相合并,经na2so4干燥,过滤,有机相旋干,上层析柱分离,使用2/8的乙酸乙酯/正己烷作为淋洗剂,得到灰白色固体产物(iiia)。nmr显示该产物具有与4-乙基-1,2,3,3a,4,8b-六氢环戊[b]吲哚一致的结构。hrms(esi,m/z):[m+h]
+
calcd for(c
13h17
n),188.1434;found,188.1336。
[0044][0045]
第二步,在500ml三口瓶中,投入第一步制备的化合物(ⅲa)(18.7g,0.1mol)、三氯氧磷(3.06g,0.02mmol)、dmf(73g,1.0mol),室温下反应6小时,旋去溶剂,旋干后柱层析[200~300目硅胶]分离提纯,用体积比为1:1的石油醚和二氯甲烷淋洗,得到固体化合物(iva)。nmr显示该产物具有与4-乙基-1,2,3,3a,4,8b-六氢环戊[b]吲哚-7-甲醛一致的结构。hrms(esi,m/z):[m+h]
+
calcd for(c
14h17
no),216.1383;found,216.1397。
[0046][0047]
第三步,在500ml两口烧瓶中,投入化合物(iva)(21.5g,0.1mol)、氰基乙酸(6.72g,0.08mol),再加入乙酸铵(3.1g,0.04mol)、乙酸(3.6g,0.06mol)和200ml甲苯,130℃反应,回流三小时。冷却到室温,有黄色固体析出,抽滤,滤饼用乙醇溶液淋洗,得黄色固体化合物(va)。nmr显示该产物具有与(e)-2-氰基-3-(4-乙基-1,2,3,3a,4,8b-六氢环戊[b]吲哚-7-基)丙烯酸一致的结构。hrms(esi,m/z):[m+h]
+
calcd for(c
17h18
n2o2),283.1441;found,283.1485。
[0048][0049]
第四步,在500ml两口烧瓶中,投入化合物(va)(28.2g,0.1mol)、乙醇(18.4g,0.4mol),再加入甲磺酸(9.6g,0.1mol)和100ml甲苯,120℃反应,回流三小时。冷却到室温,
加去离子水洗涤,蒸干溶剂,加100ml酒精,有黄色晶体析出,抽滤,滤饼用乙醇溶液淋洗,得目标化合物i-1。1h nmr(500mhz,cdcl3)δ6.68-7.84(m,4h),3.39-4.33(q,4h),2.74-3.02(t,2h),1.12-1.93(m,12h).hrms(esi,m/z):[m+h]
+
calcd for(c
19h22
n2o2),311.1754;found,311.1745。
[0050]
实施例2
[0051][0052]
第一步,将干燥的式(ii)所示化合物(15.9g,0.1mol)与对溴苯甲醚(18.7g,0.1mol)溶于230ml无水四氢呋喃中,加入干燥的四口烧瓶中,室温搅拌2h,将反应液倾倒入大量水中,二氯甲烷萃取(100ml),水洗五次(5
×
100ml),有机相合并,经na2so4干燥,过滤,有机相旋干,得到油状产物(iiib)进行下一步反应。nmr显示该产物具有与4-(4-甲氧基苯基)-1,2,3,3a,4,8b-六氢环戊[b]吲哚一致的结构。hrms(esi,m/z):[m+h]
+
calcd for(c
18h19
no),266.1539;found,266.1579。
[0053][0054]
第二步,在500ml三口瓶中,投入第一步制备的化合物(ⅲb)(26.5g,0.1mol)、三氯氧磷(3.06g,0.02mmol)、dmf(73g,1.0mol),室温下反应6小时,旋去溶剂,旋干后柱层析[200~300目硅胶],用体积比为1:1的石油醚和二氯甲烷淋洗,得到固体化合物(ivb)。nmr显示该产物具有与4-(4-甲氧基苯基)-1,2,3,3a,4,8b-六氢环戊[b]吲哚-7-甲醛一致的结构。hrms(esi,m/z):[m+h]
+
calcd for(c
19h19
no2),294.1489;found,294.1476。
[0055][0056]
第三步,在500ml两口烧瓶中,投入化合物(ivb)(29.3g,0.1mol)、氰基乙酸
(6.72g,0.08mol),再加入乙酸铵(3.1g,0.04mol)、乙酸(3.6g,0.06mol)和100ml甲苯,130℃反应,回流三小时。冷却到室温,有黄色固体析出,抽滤,滤饼用乙醇溶液淋洗,得黄色固体化合物(vb)。nmr显示该产物具有与(e)-2-氰基-3-(4-(4-甲氧基苯基)-1,2,3,3a,4,8b-六氢环戊[b]吲哚-7-基)丙烯酸一致的结构。hrms(esi,m/z):[m+h]
+
calcd for(c
22h20
n2o3),361.1547;found,361.1433。
[0057][0058]
第四步,在500ml两口烧瓶中,投入化合物(vb)(36.0g,0.1mol)、乙醇(18.4g,0.4mol),再加入三氟甲磺酸(15.0g,0.1mol)和100ml甲苯,120℃反应,回流三小时。冷却到室温,加去离子水洗涤5次(5
×
100ml),蒸干溶剂,加100ml酒精,有黄色晶体析出,抽滤,滤饼用乙醇重结晶,得目标固体化合物i-2。1h nmr(500mhz,cdcl3)δ6.79-7.88(m,8h),4.43(q,2h),2.74-3.81(m,5h),1.29-1.93(m,9h).hrms(esi,m/z):[m+h]
+
calcd for(c
24h24
n2o3),389.1860;found,389.1745。
[0059]
效果测试:将化合物i-10.1g和i-20.1g分别用10ml thf溶解稀释,分别用紫外分光光度仪测定其吸光度,所得结果如图1所示,图1是化合物i-1和化合物i-2的thf溶液(1%质量分数)的吸光度示意图。从图1可以看出,化合物i-1和化合物i-2对于波长在425nm以下的光线都具有良好的吸收作用。
[0060]
实施例3
[0061][0062]
以溴异丙烷替换实施例1中第一步反应所用的溴乙烷,其它步骤和所用试剂与实施例1相同,得到化合物i-3。1h nmr(500mhz,cdcl3)δ8.23(s,1h)6.68-7.14(m,3h),3.39-4.33(q,3h),2.74-3.02(t,2h),1.12-1.93(m,15h).hrms(esi,m/z):[m+h]
+
calcd for(c
20h24
n2o2),325.1911;found,325.1945。
[0063]
实施例4
[0064][0065]
以叔丁醇替换实施例2中第四步反应所用的乙醇,其它步骤和所用试剂与实施例2相同,得到化合物i-4。1h nmr(500mhz,cdcl3)δ6.79-7.88(m,8h),4.14(s,3h),2.74-3.81(m,2h),1.29-1.93(m,15h).hrms(esi,m/z):[m+h]
+
calcd for(c
26h28
n2o3),417.2173;found,417.2205。
[0066]
实施例5
[0067]
一种基于实施例1~4制备的吲哚类化合物作为蓝光吸收剂用于制作浇注型防蓝光基片镜片,是由表1中的重量份的组分制成:
[0068]
表1
[0069][0070]
引发剂偶氮二异丁腈的加入量占单官能基反应单体、多官能基反应单体、低聚物、引发剂和蓝光吸收剂总质量的0.1%(本实施例中加入0.1份),蓝光吸收剂化合物i-1加入量占单官能基反应单体、多官能基反应单体、低聚物、引发剂和蓝光吸收剂总质量的0.1%(本实施例中加入0.1份)。
[0071]
所述防蓝光基片镜片的制备方法包括以下步骤:
[0072]
第一步,将单官能基反应单体、多官能基反应单体、低聚物、蓝光吸收剂混合均匀加入锥形瓶中搅拌1小时,加入引发剂,在室温下抽真空30分钟,在此过程中不断搅拌混合均匀。再将温度控制在40℃,搅拌速率300r/min,静置抽真空30分钟。
[0073]
第二步,把混合均匀的第一步制备的混合物经过1.0μm的过滤器过滤,注入玻璃模具(江苏亚太光学有限公司,镜片模具)中密封,在固化炉中经过20小时从室温程序升温至95℃,程序升温曲线:
①
0-5h保持34℃;
②
5-12h匀速升至50℃;
③
12-18h匀速升至95℃;
④
18-20h保持95℃;完成一次固化获得镜片。
[0074]
第三步,将第二步获得的镜片进行开模、切边和清洗工序,在精密固化炉中110℃恒温1.5小时进行二次固化成型,得到所述防蓝光基片镜片。
[0075]
第四步,所述防蓝光基片镜片检测,检测基片的黄色指数yi(根据astm e313或hg/t3862-2006推荐的黄色指数计算方法得到,计算式为yi=100(cxx-czz)/y,其中x、y、z分别
为cie三刺激值,cx、cz为系数,本实验中取值为cx=1.28,cz=1.06),uv截止波长(以透射率达到98%的波长为值)、落球抗冲击力(根据国家标准qb/t 2506-2017测试落球冲击实验)以及抗老化测试(标准太阳光源强度照射时长,以yi值变化超过20%为计算时长终点)。
[0076]
实施例6
[0077]
一种基于实施例1~4制备的吲哚类化合物作为蓝光吸收剂用于制作浇注型防蓝光基片镜片,是由表2中的重量份的组分制成:
[0078]
表2
[0079] 成分重量份数单官能基反应单体甲基苯乙烯50份多官能基反应单体乙氧化双酚a二丙烯酸酯40份低聚物聚酯丙烯酸酯10份
[0080]
引发剂偶氮二异庚腈的加入量占单官能基反应单体、多官能基反应单体、低聚物、引发剂和蓝光吸收剂总质量的0.1%(本实施例中加入0.1份),蓝光吸收剂化合物i-1加入量占单官能基反应单体、多官能基反应单体、低聚物、引发剂和蓝光吸收剂总质量的0.05%(本实施例中加入0.05份),蓝光吸收剂化合物i-2加入量占单官能基反应单体、多官能基反应单体、低聚物、引发剂和蓝光吸收剂总质量的0.05%(本实施例中加入0.05份)。
[0081]
制备方法同实施例5。
[0082]
实施例7
[0083]
一种基于实施例1~4制备的吲哚类化合物作为蓝光吸收剂用于制作浇注型防蓝光基片镜片,是由表3中的重量份的组分制成:
[0084]
表3
[0085][0086]
引发剂偶氮二异丁腈的加入量占单官能基反应单体、多官能基反应单体、低聚物、引发剂和蓝光吸收剂总质量的0.1%(本实施例中加入0.1份),蓝光吸收剂化合物i-3加入量占单官能基反应单体、多官能基反应单体、低聚物、引发剂和蓝光吸收剂总质量的0.05%(本实施例中加入0.05份),蓝光吸收剂化合物i-4加入量占单官能基反应单体、多官能基反应单体、低聚物、引发剂和蓝光吸收剂总质量的0.05%(本实施例中加入0.05份)。
[0087]
制备方法同实施例5。
[0088]
对比例1
[0089]
现有技术中制备的基片镜片,具体成分及用量的重量份数如表4所示:
[0090]
表4
[0091][0092]
引发剂偶氮二异丁腈的加入量占单官能基反应单体、多官能基反应单体、低聚物、引发剂总质量的0.1%(本实施例中加入0.1份)。
[0093]
制备过程同实施例5,俗称白片,制作过程中区别为配方中没有添加蓝光吸收剂。
[0094]
对比例2
[0095]
现有技术中制备的基片镜片,具体成分及用量的重量份数如表5所示:
[0096]
表5
[0097] 成分重量份数单官能基反应单体甲基苯乙烯50份多官能基反应单体乙氧化双酚a二丙烯酸酯40份低聚物聚酯丙烯酸酯10份
[0098]
引发剂偶氮二异庚腈的加入量占单官能基反应单体、多官能基反应单体、低聚物、引发剂总质量的0.1%(本实施例中加入0.1份)。紫外吸收剂uv326的加入量占单官能基反应单体、多官能基反应单体、低聚物、引发剂总质量的1%(本实施例中加入1份)。
[0099]
制作过程同实施例5的防蓝光基片生产过程,同等程序升温条件,区别为配方不同。
[0100]
实施例5~7与对比例1和对比例2制备的基片镜片的性能按照实施例5给出的标准的进行测试,测试结果如表6所示:
[0101]
表6
[0102][0103]
性能测试结果表明,本发明实施例5~7使用吲哚类化合物作为蓝光吸收剂以更少的添加量(0.1%)达到对比例2的防蓝光效果,且黄色指数更低,接近于该测试条件下浇注普通镜片的水准(对比例1),抗老化性能提高,寿命延长,产品性能明显增加。
[0104]
本发明的防蓝光基片镜片的抗蓝光性能,是对可见光400-450nm波段的阻隔,该波
段为高能短波蓝光,对眼睛是有伤害的。其中列出的截止波长t%是指透射率小于2%的最大波长在410nm左右,然后呈曲线状,到450nm处渐渐透过部分蓝光,主要起到了将400-410nm部分最高能的蓝光彻底阻隔掉,而410-450nm的蓝光部分透过。这样做的一个主要原因是如果全部隔绝掉蓝光镜片彻底变黄(通过黄色指数体现),对客户平时使用增加了不舒服度。综合考虑,认为将截止波长控制在大于410nm的同时,黄色指数越低,寿命越长的镜片性能更好,实施例5~7各有优点,最优的是实施例6。
[0105]
实施例5与对比例1相比,实施例5增加了蓝光吸收剂,实施例5黄色指数比对比例1略差3.8%,差距不大,截止波长大于410nm,有抗蓝光效果,而对比例1没有该效果。落球抗冲击性能为常规试验,均合格。实施例5的抗老化性能是同等测试条件下,添加了抗蓝光吸收剂后,镜片受紫外光照射后的稳定性增加,而无蓝光吸收剂的对比例1稳定性最差,长时间受光照后黄变现象最严重。
[0106]
实施例6与对比例2相比,实施例6使用紫外吸收剂uv326,添加量为1%,实施例6与对比例2相比黄色指数大大下降,uv截止波长达到了414nm,镜片对有害蓝光的吸收效果好于对比例2,抗老化性能(或者说耐黄变性能)达到了2000h时长,测试结果是对比例2的两倍。实施例6中添加了蓝光吸收剂为0.1%,实施例6使用紫外吸收剂uv326的添加量为1%,而从截止波长数据可知,实施例6的防蓝光效果优于对比例2,说明本发明使用少量的蓝光吸收剂可以达到uv吸收剂10倍的效果,性能上更优秀。
[0107]
对比例3
[0108]
现有技术中制备的基片镜片,具体成分及用量的重量份数如表7所示:
[0109]
表7
[0110] 成分重量份数单官能基反应单体甲基苯乙烯50份多官能基反应单体乙氧化双酚a二丙烯酸酯40份低聚物聚酯丙烯酸酯10份
[0111]
引发剂偶氮二异庚腈的加入量占单官能基反应单体、多官能基反应单体、低聚物、引发剂总质量的0.1%(本实施例中加入0.1份)。紫外吸收剂uv326的加入量占单官能基反应单体、多官能基反应单体、低聚物、引发剂总质量的0.1%(本实施例中加入0.1份)。
[0112]
制备方法同实施例5。
[0113]
对比例3的性能数据:黄色指数为8.63,uv截止波长为395nm,落球冲击性能合格,抗老化性能1000h。
[0114]
与实施例6相比,对比例3的uv截止波长只到紫外区的395nm,实施例6的uv截止波长为414nm,从以上数据可以看出,对比例3的uv截止波长无法达到隔绝大部分短波蓝光的效果,即没有抗蓝光的效果。说明对比例3添加相同量的紫外吸收剂uv326与本发明实施例6添加相同量的蓝光吸收剂,对比例3并没有抗蓝光的效果,而本发明的实施例6具有抗蓝光效果。对比例3的黄色指数为8.63,实施例6的黄色指数为8.12,对比例3的黄色指数高于实施例6的黄色指数。对比例3的抗老化性能为1000h,实施例6的抗老化性能为2000h,实施例6的抗老化性能是对比例3的两倍,说明本发明实施例6添加了少量(是常规蓝光吸收剂的十分之一)蓝光吸收剂,即可达到具有抗蓝光的效果,同时,黄色指数与抗老化性能均优。
[0115]
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽
然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。