1.本发明涉及由微细化纤维素和芯粒子构成的复合粒子。
2.本技术基于2019年7月1日申请的日本特愿2019-122831号主张优先权,将其内容援引至此。
背景技术:
3.近年来,活跃地进行着将木材中的纤维素纤维微细化至其结构的至少一边达到纳米级别、作为新型的功能性材料进行利用的尝试。
4.例如,专利文献1中公开了通过对木材纤维素反复进行利用搅拌机或研磨机的机械处理,由此获得微细化纤维素纤维、即纤维素纳米纤维(以下也称作“cnf”)。记载了通过该方法获得的cnf的短轴直径为10~50nm、长轴直径为1μm~10mm。该cnf具有以钢铁1/5的重量即有5倍以上强度的优点、具有250m2/g以上的庞大的比表面积,因此期待作为树脂强化用填充物或吸附剂的利用。
5.在cnf的制造中,活跃地进行着在预先按照易于对木材中的纤维素纤维进行微细化的方式进行了化学处理后、利用家庭用混合机程度的低能量机械处理进行微细化的尝试。上述化学处理的方法并无特别限定,优选是在纤维素纤维中导入离子性官能团以易于进行微细化的方法。通过在纤维素纤维中导入离子性官能团,由于渗透压效应,溶剂易于浸入到纤维素微原纤结构之间,可以大幅度地减少纤维素原料的微细化所需要的能量。
6.上述离子性官能团的导入方法并无特别限定,例如非专利文献1中公开了使用磷酸酯化处理、选择性地对纤维素的微细纤维表面进行磷酸酯化处理的方法。专利文献2中公开了在高浓度碱水溶液中使纤维素与单氯醋酸或单氯醋酸钠反应来进行羧甲基化。还可以在高压灭菌器中使经气体化的马来酸或苯二甲酸等羧酸酐系化合物与纤维素直接反应来导入羧基。
7.另外,还报告了使用作为比较稳定的n-氧基化合物的2,2,6,6-四甲基哌啶基-1-氧自由基(tempo)作为催化剂、选择性地将纤维素的微细纤维表面氧化的方法(例如参照专利文献3)。作为催化剂使用tempo的氧化反应(tempo氧化反应)可以实施在水系、常温、常压下进行的环保型的化学改性,当适用于木材中的纤维素时,反应不会进行至结晶内部,可以仅将结晶表面的纤维素分子链所具有的醇性伯碳选择性地转变为羧基。
8.在伴随着通过tempo氧化选择性地被导入至结晶表面的羧基之间的电离的渗透压效应的作用下,可以获得在溶剂中分散成一根根的纤维素微原纤单元的纤维素单纳米纤维(以下也称作csnf)。csnf显示来自于表面羧基的高分散稳定性。有报告指出,由木材通过tempo氧化反应获得的木材来源的csnf是具有短轴直径为3nm前后、长轴直径达数十nm~数μm的高长宽比的结构体,其水分散液及成形体具有高透明性。
9.专利文献4中记载了涂布csnf分散液并干燥而获得的层叠膜具有阻气性。
10.专利文献5中记载了通过作为具有阳离子性的微细化纤维素的抗衡离子配位有机鎓阳离子的表面改性,可获得在有机溶剂中高度分散有微细化纤维素的分散液。
11.作为使用了微细化纤维素的涂饰用组合物的例子,例如在专利文献6中记载了包含tempo氧化cnf的水性涂液。该水性涂液具有良好的涂饰性,通过涂覆在锚固层上,获得具有阻隔性的层叠体。
12.专利文献7中公开了包含由于纤维素纳米纤维的长宽比高的纤维之间的缠绕或增粘特性、羧基来源的电荷的影响而分散稳定化有碳微粒的tempo氧化cnf的涂液。
13.另一方面,面对cnf的实用化,所得cnf分散液的固体成分浓度低达0.1~5%左右成为课题。例如,在想要输送微细化纤维素分散液时,由于和大量的溶剂一起进行输送,因此输送费用增大、严重影响可行性。另外,作为树脂强化用的添加剂使用时,也具有因固体成分浓度低导致的添加效率差、作为溶剂的水与树脂不相容时难以复合化的问题。进而,以含水状态使其流通时,由于还有腐败的可能,因此需要冷藏保管或防腐处理等应对,导致成本增加。
14.但是,如果单纯地通过热干燥等将微细化纤维素分散液的溶剂除去,有时微细化纤维素之间会发生凝聚、角质化或膜化,作为添加剂使用时无法稳定地表现所期待的功能。进而,由于cnf的固体成分浓度低,因此利用干燥进行的溶剂除去工序本身需要消耗巨大能量,这也成为对可行性的阻碍。
15.如此,以分散液的状态处理cnf本身就成为针对可行性的课题,强烈期待提供能够容易地处理cnf的新方式。
16.作为能够容易地处理cnf的新方式,专利文献8中记载了包含由纤维素纤维构成的被覆层和被被覆层覆盖的聚合物的复合粒子。该复合粒子中,纤维素纤维与聚合物一体化,因此可以通过过滤简单地分离,可作为粉体进行流通。粉体的再分散性也是良好的。
17.现有技术文献
18.专利文献
19.专利文献1:日本特开2010-216021号公报
20.专利文献2:国际公开第2014/088072号
21.专利文献3:日本特开2008-001728号公报
22.专利文献4:国际公开第2013/042654号
23.专利文献5:日本特开2015-101694号公报
24.专利文献6:日本专利第5928339号公报
25.专利文献7:日本专利第6020454号公报
26.专利文献8:日本特开2019-38949号公报
27.非专利文献
28.非专利文献1:noguchi y,homma i,matsubara y.complete nanofibrillation of cellulose prepared by phosphorylation.cellulose.2017;24:1295.10.1007/s10570-017-1191-3
技术实现要素:
29.发明要解决的技术问题
30.专利文献8所记载的复合粒子作为发挥如上所述cnf特性的材料是优异的,但在能够适用的聚合物的种类有限的方面仍有改善的余地。
31.当用无法适用的树脂材料形成复合粒子时,会发生收率明显降低、所得粒子的粒径分布的不均增大、由于存在于粒子表面上的cnf的量少而作为材料无法充分地发挥cnf特性等各种问题。
32.鉴于上述事实,本发明的目的在于提供处理性优异、通用性也高的纤维素纤维的复合粒子及其制造方法。
33.用于解决技术问题的手段
34.本发明的第一方式是一种复合粒子,其具备:包含至少1种聚合物的芯粒子;和具有阴离子性官能团、与芯粒子不可分地结合而配置于芯粒子表面上的微细化纤维素。
35.该复合粒子中,在微细化纤维素的至少一部分上结合有有机鎓阳离子或胺。
36.本发明的第二方式为复合粒子的制造方法。
37.该制造方法具备以下工序:在包含有机鎓化合物或胺的溶剂中对具有阴离子性的纤维素原料进行解纤、获得包含结合有有机鎓阳离子或胺的微细化纤维素的微细化纤维素分散液的第一工序;在微细化纤维素分散液中将液体状的芯粒子前体制成乳液而使其稳定化的第二工序;以及使芯粒子前体固体化而制成芯粒子、获得与芯粒子不可分地结合的微细化纤维素将芯粒子被覆而成的复合粒子的第三工序。
38.发明效果
39.根据本发明,可以提供处理性优异、通用性也高的纤维素纤维的复合粒子。
附图说明
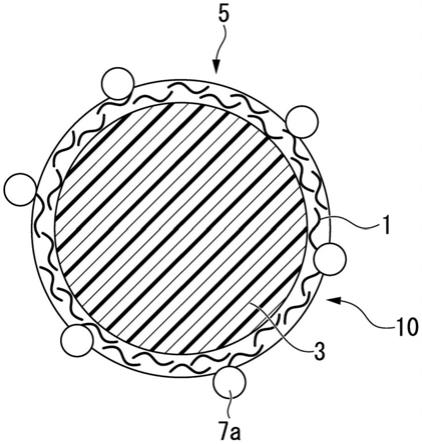
40.图1为本发明一个实施方式的复合粒子的示意图。
41.图2为表示该复合粒子的制造方法的一个过程的图。
42.图3为表示该复合粒子的制造方法的一个过程的图。
43.图4为表示该复合粒子的实施例的微细化纤维素分散液的ft-ir测定结果的曲线图。
44.图5为实施例的复合粒子的扫描电子显微镜图像。
45.图6为实施例的复合粒子的扫描电子显微镜图像。
46.图7为比较例的复合粒子的扫描电子显微镜图像。
47.图8为比较例的复合粒子的扫描电子显微镜图像。
48.图9为表示实施例的复合粒子的粒度分布的曲线图。
具体实施方式
49.以下,使用附图说明本发明的一实施方式。但是,以下说明的各图中,相互对应的部分带有同一符号,在重复部分中将后述的说明适当省略。另外,本实施方式是示例用于对本发明的技术思想进行具体化的构成,各部分的材质、形状、结构、配置、尺寸等并不限定于下述记载。本发明的技术思想在权利要求书中记载的权利要求所规定的技术范围内可以加以各种改变。
50.《复合粒子5》
51.首先,对本发明第一实施方式的微细化纤维素/芯粒子的复合粒子5进行说明。此外,芯粒子3的一个方式为树脂,也称作聚合物。
52.图1表示复合粒子5的示意图。复合粒子5具备芯粒子3、和位于芯粒子3表面上的微细化纤维素1。复合粒子5中,微细化纤维素1形成微细化纤维素层10、且与芯粒子3相结合、成为不可分的状态。
53.在微细化纤维素1的至少一部分上结合有有机鎓阳离子或胺7a作为抗衡阳离子。另外,将有机鎓化合物发生了离子化的状态记载为有机鎓离子或有机鎓阳离子。另外,这里所说的胺是指一部分或者全部发生离子化、包含铵离子。此外,之后还将有机鎓化合物或胺、或者有机鎓阳离子或铵离子的任一种表述分别记载为“有机鎓化合物/胺”、“有机鎓阳离子(或有机鎓离子)/铵离子”。
54.复合粒子5通过对使用了微细化纤维素1的o/w型皮克林乳液和乳液油滴(油相、油粒子、分散相)的液体状芯粒子前体2(以下也仅称作“液滴”)进行固体化来获得。
55.芯粒子前体2的固体化可利用各种方法进行。例如可举出作为芯粒子前体2使用具有聚合性官能团的单体(以下也称作“聚合性单体”)、在聚合过程中进行粒子形成的聚合造粒法(乳液聚合法、悬浮聚合法、种子聚合法、放射线聚合法等);由进行了微小液滴化的聚合物溶液进行粒子形成的分散造粒法(喷雾干燥法、液中固化法、溶剂蒸发法、相分离法、溶剂分散冷却法等)。
56.本实施方式中,“芯粒子前体的固体化”是指包含以下全部内容的概念:(1)将聚合性单体液滴进行聚合;(2)将聚合物液滴进行固体化;(3)将聚合性单体液滴及聚合物液滴进行固体化。
57.通过微细化纤维素1吸附在分散于连续相的亲水性溶剂中的芯粒子前体2的界面上,o/w型皮克林乳液进行稳定化。通过在维持着稳定化状态的情况下直接将乳液内部的芯粒子前体2进行固体化,可以制作以乳液为模板的复合粒子5。这里,“乳液的稳定化状态”是指即便长时间(例如12小时)静置、乳液的液滴尺寸也不会发生变化的状态。当乳液不稳定时,由于一部分液滴之间随着时间经过合而为一,液滴的粒度分布与初期相比变得更大,或者在粒度分布中发生不均,根据情况会发生油相与水相的分离。结果,所得复合粒子变得不均匀。
58.关于微细化纤维素1吸附在液滴的界面上、o/w型皮克林乳液进行稳定化的机制,认为与因吸附导致的界面能量降低所产生的作用是有关的。作为经微细化、变成亚微米级的固体粒子的微细化纤维素1通过物理的力吸附在液滴的界面上,相对于水相、形成纤维素的壁垒。一旦吸附、形成界面时,由于在解吸附中需要更大的能量,因此乳液结构进行稳定化。
59.进而还可推测以下的作用:微细化纤维素1具有双亲性,微细化纤维素1的疏水性侧吸附在具有疏水性的液滴上、亲水性朝向作为亲水性的微细化纤维素1的分散溶剂侧,因此液滴界面的稳定化有所提高。该界面上的吸附力依赖于固体粒子对油相和水相的亲和性高度,即依赖于微细化纤维素1对于芯粒子前体的亲和性和微细化纤维素1对于分散溶剂的亲和性这两者。
60.本实施方式中,通过对微细化纤维素1的一部分赋予疏水性,可提高微细化纤维素1对于液滴的亲和性,提高吸附力。作为赋予疏水性的方法,从疏水性赋予的效果高、工艺成本有利的方面出发,优选对于具有阴离子性官能团的微细化纤维素1使用有机鎓化合物/胺7、使抗衡离子为有机鎓离子/铵离子7a的方法。
61.通过使用一部分经疏水化的微细化纤维素1来制作复合粒子5,乳液的稳定性提高。结果,能够适用的芯粒子前体的材料种类大幅度地增加,可以制作能够与和用途等相应的需求规格对应的多种复合粒子5。
62.本实施方式中,表示芯粒子3与微细化纤维素1的结合状态的“不可分”是指即使下述那样的操作反复进行后,微细化纤维素1和芯粒子3也不会分离,可保持微细化纤维素1对芯粒子3的被覆状态,上述的操作是:通过对包含复合粒子5的分散液进行离心分离处理后将上清除去、进而添加溶剂进行再分散来对复合粒子5进行纯化、洗涤的操作;或者通过使用了滤膜的过滤洗涤反复用溶剂进行洗涤的操作。
63.微细化纤维素1对芯粒子3的被覆状态可以通过利用扫描型电子显微镜(sem)的复合粒子5的表面观察来确认。微细化纤维素1与芯粒子3不可分地结合的详细机制虽不清楚,但认为是由于复合粒子5是以被微细化纤维素1稳定化了的o/w型乳液为模板制作而成,因此当在微细化纤维素1接触于乳液内部的液滴的状态下进行液滴的固体化时,微细化纤维素1的一部分保持着位于液滴的状态被固定化,最终芯粒子3与微细化纤维素1不可分地进行结合。
64.o/w型乳液也称作水包油滴型(oil-in-water),是以水为连续相,油以油滴(油粒子)的形式分散在其中。
65.复合粒子5由于是以被微细化纤维素1稳定化了的o/w型乳液为模板制作而成,因此其特征之一在于成为源于o/w型乳液的接近于圆球的形状。在典型的复合粒子5中,由微细化纤维素1形成的微细化纤维素层10以较均匀的厚度形成在圆球状的芯粒子3的表面上。
66.微细化纤维素层10的平均厚度通过用切片机对用包埋树脂固定的复合粒子5进行切削后进行sem观察,在图像上的随机100处测定图像中的复合粒子5的截面图像中的微细化纤维素层10的厚度、并算出其算术平均值,从而获得。
67.微细化纤维素层10的厚度是均匀的,这也是复合粒子5的一个特征。微细化纤维素层10的厚度值的变异系数(从上述100处随机抽取的30处的标准偏差)优选为0.5以下、更优选为0.4以下。
68.本实施方式的微细化纤维素1并无特别限定,优选在结晶表面上具有阴离子性官能团,该阴离子性官能团的含量在纤维素每1g中为0.1mmol以上且5.0mmol以下。
69.进而,微细化纤维素1优选是微原纤结构来源的纤维形状。具体地说,微细化纤维素1优选是纤维状,数均短轴直径为1nm以上且1000nm以下、数均长轴直径为50nm以上,且数均长轴直径为数均短轴直径的5倍以上。另外,微细化纤维素1的结晶结构优选是纤维素i型。
70.《复合粒子5的制造方法》
71.对本实施方式的复合粒子5的制造方法进行说明。本实施方式的复合粒子的制造方法具备以下工序:在包含有机鎓化合物/胺7的溶剂中对具有阴离子性的纤维素原料6进行解纤、获得微细化纤维素分散液的工序(第一工序);在微细化纤维素分散液中将液体状的芯粒子前体2制成乳液、使其稳定化的工序(第二工序);以及使芯粒子前体2固体化、获得被微细化纤维素1被覆的复合粒子的工序(第三工序)。
72.通过上述制造方法获得的复合粒子5作为分散体获得。将溶剂从分散体中除去时,获得处理性良好的复合粒子5的干燥固形物。溶剂的除去方法并无特别限定,例如可以示例
通过离心分离法或过滤法将溶剂除去的方法;在烘箱等中使溶剂气化而除去的方法。此时,所得复合粒子5的干燥固形物不会变为膜状或凝聚体状,而是成为细腻的粉体。其原因虽还不确定,但认为其原因之一在于,为包含复合粒子5的分散液时,由于是表面固定化有微细化纤维素1的大致圆球状的复合粒子,因此即便是将溶剂除去、微细化纤维素1之间也不会凝聚,相邻的复合粒子间仅进行点接触。由于复合粒子5不会发生凝聚,因此也易于将作为干燥粉体获得的复合粒子5再分散在溶剂中,再分散后也显示来自结合于表面的微细化纤维素1的分散稳定性。
73.复合粒子5的干燥粉体是以几乎不含溶剂、能够进一步再分散于溶剂中为优点的干燥固形物,具体地说可以使固体成分率为80%以上、进一步可以为90%以上、进一步可以为95%以上。
74.复合粒子5的分散体由于将溶剂几乎除去是容易的,因此从输送费用的削减、防止腐败、添加率提高、与树脂的混炼效率提高的观点出发,可获得优选的效果。此外,即便是在利用干燥处理使固体成分率为80%以上时,由于微细化纤维素1易于吸湿,因此会将空气中的水分吸附而使固体成分率经时地降低,在保管过程中有时可能变为80%以下。但是,考虑到以复合粒子5作为干燥粉体易于获得、可进一步使其再分散为优点的本发明技术思想时,只要是经过使包含复合粒子5的干燥粉体的固体成分率达到80%以上的工序而获得的干燥固形物,则应该说是包含在本发明的技术范围内的。
75.以下,详细地说明制造方法的各工序。
76.(第一工序)
77.将本实施方式的第一工序示于图2中。
78.第一工序是在包含有机鎓化合物/胺7的溶剂中对具有阴离子性的纤维素原料6进行解纤、获得微细化纤维素分散液的工序。
79.首先,如图2所示,将具有各种阴离子性的纤维素原料6与有机鎓化合物/胺7混合。由此,有机鎓化合物/胺7中所含的有机鎓阳离子/铵离子7a与纤维素原料6结合,对纤维素原料6的一部分赋予疏水性。
80.具有阴离子性的纤维素原料6还可以形成以金属离子为代表的阳离子性物质作为抗衡离子的盐,但不含阳离子物质时,能够抑制副产物的生成、疏水化的效果优异,因此优选不含阳离子性物质。
81.纤维素原料6中的阳离子性物质的含量可以利用各种分析方法研究。例如,作为简单的方法,可以示例使用了电子束微量分析器的epma法或利用荧光x射线分析法的元素分析等。作为从具有阴离子性的纤维素原料6中除去阳离子性物质的方法,可以示例在酸性下反复洗涤纤维素原料6之后、利用纯水反复进行水洗的纯化方法。
82.作为有机铵化合物/胺7的结合量,优选相对于纤维素原料6所含的阴离子性官能团为0.8当量以上且2当量以下。特别是,当结合量为1当量以上且1.8当量以下时,没有必要添加大量的有机鎓化合物/胺7,更为优选。即便有机鎓化合物/胺7的结合量小于0.8当量,也可以某种程度地使纤维素原料6分散,但在分散处理时需要长时间、高能量,所得纤维的纤维径变大、均质性降低。另一方面,当超过2当量时,由于有机鎓化合物/胺7的过量添加,有发生纤维素原料6分解或对分散介质的亲和性降低的情况,从而不优选。
83.有机鎓化合物/胺7的种类可以使用1种,也可以混合使用2种以上。特别是,还可以
100.n——r4r5r6···
(3)
101.nh
+
——r4r5r6···
(3)’102.上述结构式中,r1~r6是烃基、含杂原子的烃基中的任一种。
103.作为伯胺、仲胺、叔胺,可以示例甲基胺、乙基胺、丙基胺、丁基胺、正辛基胺、二甲基胺、二乙基胺、二丙基胺、二丁基胺、己基胺、2-乙基己基胺、二己基胺、三甲基胺、三乙基胺、三丙基胺、三丁基胺、三辛基胺、三己基胺、二辛基胺、十二烷基胺、硬脂基胺、油胺等。
104.使用具有阴离子性的纤维素原料6和有机鎓化合物/胺7获得的悬浮液与使用以金属离子为抗衡离子的无机碱的情况相比,能够以更低能量、更短时间进行分散处理,且最终获得的分散液的均质性也更高。认为其原因在于,使用了有机鎓化合物/胺7时,由于纤维素原料6具有的阴离子性部位的抗衡离子的离子径更大,因此在分散介质中将微细化纤维素纤维之间进一步拉开的效果更大。进而,作为分散液包含有机鎓化合物/胺7时,与无机碱相比,可以降低分散液的粘度和触变性,在分散处理的容易性及之后的处理中变得有利。进而,通过离子键与有机鎓化合物/胺7发生相互作用的微细化纤维素1通过基于有机鎓离子/铵离子7a的位阻斥力或疏水化作用,亲水性降低。由此,在后述的第二工序中,对液状的芯粒子前体2的乳液液滴的亲和性提高、液滴的稳定性提高。
105.接着,对悬浮液实施物理性解纤处理,对纤维素原料6进行微细化。作为物理性解纤处理的方法并无特别限定,可以示例高压匀浆机、超高压匀浆机、球磨机、辊磨机、切碎机、行星式研磨机、喷磨机、磨碎机、研磨机、榨汁机、高速搅拌机、超声波匀浆机、微射流均质机、水中对向冲撞等机械处理。通过进行物理性解纤处理,将悬浮液中的纤维素原料6微细化,可以获得其结构的至少一边被微细化至纳米级别的微细化纤维素1的分散液。通过改变物理性解纤处理的时间或次数,可以调整分散液所含的微细化纤维素1的数均短轴直径及数均长轴直径。
106.由以上,获得图2所示的微细化纤维素1的分散液4。将微细化纤维素1微细化至其结构的至少一边达到纳米级别。
107.微细化纤维素1由于是微原纤结构来源的纤维形状,因此作为本实施方式的制造方法中使用的微细化纤维素1,优选处于以下所示范围内的纤维形状者。即,纤维状的微细化纤维素1只要是就短轴直径而言是数均短轴直径为1nm以上且1000nm以下即可、优选为2nm以上且500nm以下即可。这里,当数均短轴直径小于1nm时,无法取高结晶性的刚直的微细化纤维素纤维结构,无法实施乳液的稳定化和以乳液为模板的聚合反应。另一方面,数均短轴直径超过1000nm时,由于在乳液稳定化中尺寸变得过大,因此变得难以控制所得复合粒子5的尺寸或形状。
108.微细化纤维素1的数均长轴直径并无特别限定,优选为数均短轴直径的5倍以上即可。当数均长轴直径小于数均短轴直径的5倍时,由于无法充分地控制复合粒子5的尺寸或形状,因此不优选。
109.微细化纤维素1的数均短轴直径是利用透射型电子显微镜观察及原子间力显微镜观察测定100根纤维的短轴直径(最小径)、作为其平均值求得的。微细化纤维素1的数均长轴直径是利用透射型电子显微镜观察及原子间力显微镜观察测定100根纤维的长轴直径(最大径)、作为其平均值求得的。
110.分散液4中,有机鎓化合物/胺7结合在微细化纤维素1上,显示良好的分散。分散液
4在之后的工序中可以直接使用或者进行稀释、浓缩等后作为o/w型乳液的稳定化剂进行使用。
111.构成微细化纤维素1的纤维素的种类或结晶结构也无特别限定。具体地说,作为由纤维素i型结晶构成的原料,例如除了木材系天然纤维素之外,还可以使用棉短绒、竹、麻、甘蔗渣、洋麻、细菌纤维素、海鞘纤维素、法囊藻纤维素等非木材系天然纤维素。进而,还可以使用由纤维素ii型结晶构成的人造丝纤维、铜氨纤维为代表的再生纤维素。出于材料调配容易的理由,优选以木材系天然纤维素为原料。作为木材系天然纤维素并无特别限定,可以使用针叶树纸浆或者阔叶树纸浆、废纸纸浆等通常在纤维素纳米纤维的制造中使用的材料。出于纯化及微细化容易的理由,优选针叶树纸浆。
112.进而,优选对纤维素原料6进行化学改性。更具体地说,优选将阴离子型官能团导入至纤维素原料6的结晶表面。其原因在于,通过在纤维素原料6的结晶表面导入阴离子性官能团,由于渗透压效应、溶剂易于浸入纤维素结晶之间,纤维素原料6的微细化易于进行。
113.导入至纤维素的结晶表面的阴离子性官能团的种类或导入方法并无特别限定,优选羧基或磷酸基。出于对纤维素结晶表面的选择性导入容易的理由,优选羧基。
114.在纤维素的结晶表面导入羧基的方法并无特别限定。具体地说,例如可以通过在高浓度碱水溶液中使纤维素与单氯乙酸或单氯乙酸钠反应来进行羧甲基化。另外,还可以在高压灭菌器中使经气化的马来酸或苯二甲酸等羧酸酐系化合物与纤维素直接反应来导入羧基。进而,还可以使用在水系的较温和的条件下、在尽量保持结构的同时、在以对于醇性伯碳的氧化的选择性高的tempo为首的n-氧基化合物的存在下、使用共氧化剂的手法。为了羧基导入部位的选择性及降低环境负荷,更优选使用了n-氧基化合物的氧化。
115.这里,作为n-氧基化合物,可举出tempo(2,2,6,6-四甲基哌啶基-1-氧自由基)、2,2,6,6-四甲基-4-羟基哌啶-1-氧基、4-甲氧基-2,2,6,6-四甲基哌啶-n-氧基、4-乙氧基-2,2,6,6-四甲基哌啶-n-氧基、4-乙酰胺-2,2,6,6-四甲基哌啶-n-氧基等。其中,优选反应性高的tempo。n-氧基化合物的使用量只要是作为催化剂的量即可,并无特别限定。通常相对于进行氧化处理的木材系天然纤维素的固体成分为0.01~5.0质量%左右。
116.作为使用了n-氧基化合物的氧化方法,例如可举出使木材系天然纤维素分散在水中、在n-氧基化合物的共存下进行氧化处理的方法。此时,优选与n-氧基化合物一起并用共氧化剂。此时,在反应体系内,n-氧基化合物依次被共氧化剂氧化而生成氧代铵盐,通过上述氧代铵盐,纤维素被氧化。通过该氧化处理,即便是在温和的条件下,氧化反应也会顺畅地进行,羧基的导入效率提高。在温和的条件下进行氧化处理时,易于维持纤维素的结晶结构。
117.作为共氧化剂,只要是卤素、次卤酸、亚卤酸或高卤酸、或者它们的盐、卤素氧化物、氮氧化物、过氧化物等能够推进氧化反应者,则可以使用任一种氧化剂。从获得容易或反应性出发,优选次氯酸钠。上述共氧化剂的使用量只要是能够促进氧化反应的量即可,并无特别限定。通常相对于进行氧化处理的木材系天然纤维素的固体成分为1~200质量%左右。
118.另外,还可以与n-氧基化合物及共氧化剂一起进一步并用选自溴化物或碘化物中的至少1种化合物。由此,可以使氧化反应顺畅地进行,可以改善羧基的导入效率。作为这种化合物,优选溴化钠或溴化锂,从成本或稳定性出发,优选溴化钠。化合物的使用量只要是
能够促进氧化反应的量即可,并无特别限定。通常相对于进行氧化处理的木材系天然纤维素的固体成分为1~50质量%左右。
119.氧化反应的反应温度优选为4~80℃、更优选为10~70℃。小于4℃时,试剂的反应性降低、反应时间变长。超过80℃时,副反应促进、试样发生低分子化、高结晶性的刚直的微细化纤维素纤维结构破坏,无法作为o/w型乳液的稳定化剂使用。
120.另外,氧化处理的反应时间可以考虑反应温度、所希望的羧基量等来适当设定,虽无特别限定,通常为10分钟~5小时左右。
121.氧化反应时的反应体系的ph并无特别限定,优选为9~11。ph为9以上时,可以高效地使反应进行。ph超过11时,有副反应进行、促进试样分解的危险。另外,在氧化处理中,随着氧化进行而生成羧基,因此体系内的ph会降低,因而在氧化处理中,优选将反应体系的ph保持在9~11。作为将反应体系的ph保持在9~11的方法,可举出对应于ph的降低而添加碱水溶液的方法。
122.作为碱水溶液,可举出氢氧化钠水溶液、氢氧化锂水溶液、氢氧化钾水溶液、氨水溶液、氢氧化四甲基铵水溶液、氢氧化四乙基铵水溶液、氢氧化四丁基铵水溶液、氢氧化苄基三甲基铵水溶液等有机鎓化合物的水溶液等。从成本等方面出发,优选氢氧化钠水溶液。
123.利用n-氧基化合物进行的氧化反应通过在反应体系中添加醇而使其停止。此时,优选反应体系的ph保持在上述范围内。作为所添加的醇,由于会使反应迅速结束,因此优选甲醇、乙醇、丙醇等低分子量的醇,出于通过反应生成的副产物的安全性等理由,特别优选乙醇。
124.氧化处理后的反应液也可以直接供至微细化工序,但为了将n-氧基化合物等催化剂、杂质等除去,优选将反应液所含的氧化纤维素回收、利用洗涤液进行洗涤。氧化纤维素的回收可以通过使用玻璃过滤器或者20μm孔径尼龙筛的过滤等公知方法来实施。作为氧化纤维素的洗涤中使用的洗涤液,优选纯水。
125.当对所得tempo氧化纤维素进行解纤处理时,可获得具有3nm的均匀纤维宽度的纤维素单纳米纤维(csnf)。使用csnf作为复合粒子5的微细化纤维素1的原料时,由于其均匀的结构、所得o/w型乳液的粒径也易于变得均匀。
126.如上所述,本实施方式中使用的csnf可以通过对纤维素原料进行氧化的工序和进行微细化而制成分散液的工序来获得。另外,作为导入至csnf中的羧基的含量,优选为0.1mmol/g以上且5.0mmol/g以下、更优选为0.5mmol/g以上且2.0mmol/g以下。这里,羧基量小于0.1mmol/g时,在纤维素微原纤之间、渗透压效应所产生的溶剂进入作用不发挥,因此难以将纤维素进行微细化而难以使其均匀地分散。另外,超过5.0mmol/g时,由于伴随化学处理的副反应,纤维素微原纤发生低分子化,因此不会成为高结晶性的刚直的微细化纤维素纤维结构,无法作为o/w型乳液的稳定化剂使用。
127.分散液4也可根据需要在不损害本发明效果的范围内含有除纤维素及在ph调节中使用的成分以外的其它成分。作为其它的成分并无特别限定,可以根据复合粒子5的用途等从公知的添加剂中适当选择。具体地说,可举出烷氧基硅烷等有机金属化合物或其水解物、无机层状化合物、无机针状矿物、消泡剂、无机系粒子、有机系粒子、润滑剂、抗氧化剂、防静电剂、紫外线吸收剂、稳定剂、磁性粉、取向促进剂、增塑剂、交联剂、磁性体、药品、农药、香料、粘接剂、酶、颜料、染料、除臭剂、金属、金属氧化物、无机氧化物等。
128.(第二工序)
129.第二工序是在第一工序中获得的微细化纤维素1的分散液4中,用微细化纤维素1将液体状的芯粒子前体2的表面被覆,以乳液的形式使其稳定化的工序。
130.具体地说,是在第一工序中获得的分散液4中添加液体状的芯粒子前体2,使芯粒子前体2作为液滴分散在分散液4中,用微细化纤维素1将液滴的表面被覆,如图3左侧所示,制作被微细化纤维素层10稳定化了的o/w型乳液的工序。
131.作为制作o/w型乳液的方法并无特别限定,可以使用一般的乳化处理,例如各种匀浆机处理或机械搅拌处理,具体地说可举出高压匀浆机、超高压匀浆机、万能匀浆机、球磨机、辊磨机、切碎机、行星式研磨机、喷磨机、磨碎机、研磨机、榨汁机、高速搅拌机、超声波匀浆机、微射流均质机、水中对向冲撞、涂料搅拌器等机械处理。另外,还可以组合使用多个机械处理。
132.例如使用超声波匀浆机时,对第一工序中获得的微细化纤维素分散液添加聚合性单体制作混合溶剂,在混合溶剂中插入超声波匀浆机的前端,实施超声波处理。作为超声波匀浆机的处理条件并无特别限定,例如频率一般为20khz以上、输出功率一般为10w/cm2以上。处理时间也无特别限定,通常为10秒钟~1小时左右。
133.通过上述超声波处理,芯粒子前体2的液滴分散在分散液4中、乳液化进行,进而微细化纤维素1选择性地吸附在液滴与分散液4的液/液界面上,从而形成液滴被微细化纤维素1被覆、作为o/w型乳液而稳定化了的结构。如此,固体物吸附在液/液界面上而稳定化了的乳液在学术上被称作“皮克林乳液”。如上所述,通过微细化纤维素纤维形成皮克林乳液的机制虽不确定,但认为是由于纤维素在其分子结构中具有来自于羟基的亲水性位点和来自烃基的疏水性位点,从而显示双亲性,由于双亲性而吸附在疏水性单体和亲水性溶剂的液/液界面上。
134.o/w型乳液结构可以通过光学显微镜观察进行确认。o/w型乳液的粒径尺寸并无特别限定,通常为0.1μm~1000μm左右。
135.o/w型乳液结构中,形成于液滴的表层的微细化纤维素层10的厚度并无特别限定,通常为3nm~1000nm左右。微细化纤维素层10的厚度例如可以使用cryo tem测量。
136.作为第二工序中能够使用的液体状的芯粒子前体2的材质,只要是与水不互溶且为(1)为具有聚合性官能团的单体(聚合性单体)、可通过聚合反应形成固体聚合物的材质;(2)为热塑性聚合物、通过加热而熔融成液体状、发生相转移、在室温下变为固体的材质;及(3)为非固化性聚合物、通过溶剂而溶解成液体状、通过溶剂除去而在室温下变为固体的材质中的任一种,则无特别限定。
137.就(1)而言,聚合性单体具有至少1个聚合性官能团。具有一个聚合性官能团的聚合性单体也称作单官能单体。另外,具有两个以上聚合性官能团的聚合性单体也称作多官能单体。作为聚合性单体的种类并无特别限定,例如可举出(甲基)丙烯酸系单体、乙烯基系单体等。另外,还可以使用具有环氧基或氧杂环丁烷结构等环状醚结构的聚合性单体(例如ε-己内酯等)。
138.此外,“(甲基)丙烯酸酯”的表述是指包含“丙烯酸酯”和“甲基丙烯酸酯”这两者。
139.作为单官能的(甲基)丙烯酸系单体,例如可举出(甲基)丙烯酸2-羟基乙酯、(甲基)丙烯酸2-羟基丙酯、(甲基)丙烯酸2-羟基丁酯、(甲基)丙烯酸正丁酯、(甲基)丙烯酸异
丁酯、(甲基)丙烯酸叔丁酯、(甲基)丙烯酸缩水甘油酯、丙烯酰基吗啉、n-乙烯基吡咯烷酮、丙烯酸四氢糠酯、(甲基)丙烯酸环己酯、(甲基)丙烯酸2-乙基己酯、(甲基)丙烯酸异冰片酯、(甲基)丙烯酸异癸酯、(甲基)丙烯酸月桂酯、(甲基)丙烯酸十三烷基酯、(甲基)丙烯酸鲸蜡酯、(甲基)丙烯酸硬脂酯、(甲基)丙烯酸苄酯、(甲基)丙烯酸2-乙氧基乙酯、(甲基)丙烯酸3-甲氧基丁酯、乙基卡必醇(甲基)丙烯酸酯、磷酸(甲基)丙烯酸酯、环氧乙烷改性磷酸(甲基)丙烯酸酯、(甲基)丙烯酸苯氧酯、(甲基)丙烯酸环氧乙烷改性苯氧酯、(甲基)丙烯酸环氧丙烷改性苯氧酯、壬基苯酚(甲基)丙烯酸酯、环氧乙烷改性壬基苯酚(甲基)丙烯酸酯、环氧丙烷改性壬基苯酚(甲基)丙烯酸酯、甲氧基二乙二醇(甲基)丙烯酸酯、甲氧基聚乙二醇(甲基)丙烯酸酯、甲氧基丙二醇(甲基)丙烯酸酯、2-(甲基)丙烯酰氧基乙基-2-羟基丙基邻苯二甲酸酯、2-羟基-3-苯氧基丙基(甲基)丙烯酸酯、2-(甲基)丙烯酰氧基乙基氢化邻苯二甲酸酯、2-(甲基)丙烯酰氧基丙基氢化邻苯二甲酸酯、2-(甲基)丙烯酰氧基丙基六氢邻苯二甲酸酯、2-(甲基)丙烯酰氧基丙基四氢邻苯二甲酸酯、(甲基)丙烯酸二甲基氨基乙酯、(甲基)丙烯酸三氟乙酯、(甲基)丙烯酸四氟丙酯、(甲基)丙烯酸六氟丙酯、(甲基)丙烯酸八氟丙酯、由2-金刚烷及金刚烷二醇衍生的具有1价单(甲基)丙烯酸酯的金刚烷基丙烯酸酯等金刚烷衍生物单(甲基)丙烯酸酯等。
140.作为二官能的(甲基)丙烯酸系单体,例如可举出乙二醇二(甲基)丙烯酸酯、二乙二醇二(甲基)丙烯酸酯、丁二醇二(甲基)丙烯酸酯、己二醇二(甲基)丙烯酸酯、壬二醇二(甲基)丙烯酸酯、乙氧基化己二醇二(甲基)丙烯酸酯、丙氧基化己二醇二(甲基)丙烯酸酯、二乙二醇二(甲基)丙烯酸酯、聚乙二醇二(甲基)丙烯酸酯、三丙二醇二(甲基)丙烯酸酯、聚丙二醇二(甲基)丙烯酸酯、新戊二醇二(甲基)丙烯酸酯、乙氧基化新戊二醇二(甲基)丙烯酸酯、三丙二醇二(甲基)丙烯酸酯、羟基新戊酸新戊二醇二(甲基)丙烯酸酯等二(甲基)丙烯酸酯等。
141.作为三官能以上的(甲基)丙烯酸系单体,例如可举出三羟甲基丙烷三(甲基)丙烯酸酯、乙氧基化三羟甲基丙烷三(甲基)丙烯酸酯、丙氧基化三羟甲基丙烷三(甲基)丙烯酸酯、三(2-羟基乙基异氰脲酸酯)三(甲基)丙烯酸酯、甘油三(甲基)丙烯酸酯等三(甲基)丙烯酸酯,季戊四醇三(甲基)丙烯酸酯、二季戊四醇三(甲基)丙烯酸酯、二(三羟甲基丙烷)三(甲基)丙烯酸酯等三官能的(甲基)丙烯酸酯化合物;季戊四醇四(甲基)丙烯酸酯、二(三羟甲基丙烷)四(甲基)丙烯酸酯、二季戊四醇四(甲基)丙烯酸酯、二季戊四醇五(甲基)丙烯酸酯、二(三羟甲基丙烷)五(甲基)丙烯酸酯、二季戊四醇六(甲基)丙烯酸酯、二(三羟甲基丙烷)六(甲基)丙烯酸酯等三官能以上的多官能(甲基)丙烯酸酯化合物、或将这些(甲基)丙烯酸酯的一部分用烷基或ε-己内酯取代了的多官能(甲基)丙烯酸酯化合物等。
142.作为单官能的乙烯基系单体,例如优选乙烯基醚系、乙烯基酯系、芳香族乙烯基系、特别是苯乙烯及苯乙烯系单体等在常温下与水不互溶的液体。
143.作为单官能乙烯基系单体中的(甲基)丙烯酸酯,可举出(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯、(甲基)丙烯酸丁酯、(甲基)丙烯酸叔丁酯、(甲基)丙烯酸2-乙基己酯、(甲基)丙烯酸月桂酯、(甲基)丙烯酸烷基酯、(甲基)丙烯酸十三烷基酯、(甲基)丙烯酸硬脂酯、(甲基)丙烯酸环己酯、(甲基)丙烯酸苄酯、(甲基)丙烯酸异冰片酯、(甲基)丙烯酸缩水甘油酯、(甲基)丙烯酸四氢糠酯、(甲基)丙烯酸烯丙酯、(甲基)丙烯酸二乙基氨基乙酯、(甲基)丙烯酸三氟乙酯、(甲基)丙烯酸七氟癸酯、(甲基)丙烯酸二环戊烯酯、(甲基)丙烯酸二环戊
烯基氧基乙酯、(甲基)丙烯酸三环癸酯等。
144.另外,作为单官能芳香族乙烯基系单体,可举出苯乙烯、α-甲基苯乙烯、邻甲基苯乙烯、间甲基苯乙烯、对甲基苯乙烯、乙基苯乙烯、异丙烯基甲苯、异丁基甲苯、叔丁基苯乙烯、乙烯基萘、乙烯基联苯、1,1-二苯基乙烯等。
145.作为多官能的乙烯基系单体,可举出二乙烯基苯等具有不饱和键的多官能团。优选常温下与水不互溶的液体。
146.例如,作为多官能性乙烯基系单体,具体地可举出(1)二乙烯基苯、1,2,4-三乙烯基苯、1,3,5-三乙烯基苯等二乙烯基类;(2)乙二醇二甲基丙烯酸酯、二乙二醇二甲基丙烯酸酯、三乙二醇二甲基丙烯酸酯、聚乙二醇二甲基丙烯酸酯、1,3-丙二醇二甲基丙烯酸酯、1,4-丁二醇二甲基丙烯酸酯、1,6-己二醇二甲基丙烯酸酯、新戊二醇二甲基丙烯酸酯、二丙二醇二甲基丙烯酸酯、聚丙二醇二甲基丙烯酸酯、2,2-双(4-甲基丙烯酰氧基二乙氧基苯基)丙烷等二甲基丙烯酸酯类;(3)三羟甲基丙烷三甲基丙烯酸酯、三羟乙基乙烷三甲基丙烯酸酯等三甲基丙烯酸酯类;(4)乙二醇二丙烯酸酯、二乙二醇二丙烯酸酯、三乙二醇二丙烯酸酯、聚乙二醇二丙烯酸酯、1,3-二丙二醇二丙烯酸酯、1,4-二丁二醇二丙烯酸酯、1,6-己二醇二丙烯酸酯、新戊二醇二丙烯酸酯、二丙二醇二丙烯酸酯、聚丙二醇二丙烯酸酯、2,2-双(4-丙烯酰氧基丙氧基苯基)丙烷、2,2-双(4-丙烯酰氧基二乙氧基苯基)丙烷等二丙烯酸酯类;(5)三羟甲基丙烷三丙烯酸酯、三羟乙基乙烷三丙烯酸酯等三丙烯酸酯类;(6)四羟甲基甲烷四丙烯酸酯等四丙烯酸酯类;(7)其它例如四亚甲基双(乙基富马酸酯)、六亚甲基双(丙烯酰胺)、三烯丙基氰脲酸酯、三烯丙基异氰脲酸酯。
147.例如作为官能性苯乙烯系单体,具体地可举出二乙烯基苯、三乙烯基苯、二乙烯基甲苯、二乙烯基萘、二乙烯基二甲苯、二乙烯基联苯、双(乙烯基苯基)甲烷、双(乙烯基苯基)乙烷、双(乙烯基苯基)丙烷、双(乙烯基苯基)丁烷等。
148.近年来,考虑到环境,强烈期待具有在使用后被自然界的微生物分解、最终可以变为水和二氧化碳的生物降解性的粉体。
149.本实施方式的芯粒子3中可以使用生物降解性材料。作为生物降解性材料,只要是具有生物降解性、不溶解于水的材料,则无特别限定,具体地说可以示例醋酸纤维素、醋酸丁酸纤维素、醋酸丙酸纤维素等醋酸纤维素衍生物,壳多糖、壳聚糖等多糖类,聚乳酸、乳酸与其它羟基羧酸的共聚物等聚乳酸类;聚琥珀酸丁二醇酯、聚琥珀酸乙二醇酯、聚己二酸丁二醇酯等二元酸聚酯类,聚己内酯、己内酯与羟基羧酸的共聚物等聚己内酯类;聚羟基丁酸酯、聚羟基丁酸酯与羟基羧酸的共聚物等聚羟基丁酸酯类;聚羟基丁酸、聚羟基丁酸与其它羟基羧酸的共聚物等脂肪族聚酯类;聚氨基酸类、聚酯聚碳酸酯类、松香等天然树脂等。这些化合物可以单独使用或者并用2种以上来使用。
150.另外,除这些以外,还可以使用具有至少1个以上聚合性官能团的聚醚树脂、聚酯树脂、聚氨酯树脂、环氧树脂、醇酸树脂、螺缩醛树脂、聚丁二烯树脂、聚巯基聚烯树脂等,并不特别限定该材料。
151.上述各种聚合性单体可以单独使用,也可以组合使用2种以上。
152.就上述(2)而言,作为热塑性聚合物,优选熔点为40℃以上且80℃以下。熔点低于40℃时,在室温下作为固体难以维持形状、极端地限制了使用环境,因此不优选。另一方面,当熔点超过80℃时,从制造工序上来说,在微细化纤维素分散液中难以维持熔融状态,因此
不优选。更优选熔点为45℃以上且75℃以下。另外,熔点以上的熔体流动速率(mfr)优选为10以上。mfr小于10时,在前述的乳化处理中,需要大量的乳化能量,因此不优选。
153.就上述(3)而言,作为非固化性聚合物,是溶解在除水之外的溶剂中、具有液体状的物质。这里,作为溶解非固化性聚合物的溶剂,优选20℃下的水溶解度是相对于水1l为20g以上且2000g以下。小于20g时,包含溶剂的液滴与微细化纤维素1的亲和性低、由微细化纤维素1产生的乳液稳定化效果降低。另一方面,大于2000g时,由于在微细化纤维素分散液中的溶剂的扩散速度快,因此液滴无法维持形状。结果会损害微细化纤维素1产生的液滴被覆效果。
154.热塑性聚合物及非固化性聚合物只要不损害本实施方式的功能,则其材料并无限定。例如,可以使用以上述各种单官能单体、或者以具有环氧基或氧杂环丁烷结构等环状醚结构的聚合性单体作为起始物质获得的聚合物、或者聚醚树脂、聚酯树脂、聚氨酯树脂、环氧树脂、醇酸树脂、螺缩醛树脂、聚丁二烯树脂、聚巯基聚烯树脂等。
155.第二工序中的分散液4与液体状芯粒子前体2的重量比并无特别限定,相对于100质量份的分散液4,芯粒子前体2优选为1质量份以上且50质量份以下。芯粒子前体2为1质量份以下时,由于复合粒子5的收量降低,因此不优选,超过50质量份时,难以用微细化纤维素1均匀地将液滴被覆,从而不优选。
156.作为液体状的芯粒子前体2使用聚合性单体时,还可以预先包含聚合引发剂。作为通常的聚合引发剂,可以示例有机过氧化物或偶氮聚合引发剂等自由基引发剂等。
157.作为有机过氧化物,可以示例过氧缩酮、过氧化氢、二烷基过氧化物、二酰基过氧化物、过氧化碳酸酯、过氧化酯等。
158.作为偶氮聚合引发剂,可以示例advn、aibn等。
159.例如为2,2-偶氮双(异丁腈)(aibn)、2,2-偶氮双(2-甲基丁腈)(ambn)、2,2-偶氮双(2,4-二甲基戊腈)(advn)、1,1-偶氮双(1-环己烷碳腈)(achn)、2,2-偶氮双异丁酸二甲酯(maib)、4,4-偶氮双(4-氰基戊酸)(acva)、1,1-偶氮双(1-乙酰氧基-1-苯基乙烷)、2,2-偶氮双(2-甲基丁基酰胺)、2,2-偶氮双(4-甲氧基-2,4-二甲基戊腈)、2,2-偶氮双(2-甲基脒基丙烷)二盐酸盐、2,2-偶氮双[2-(2-咪唑啉-2-基)丙烷]、2,2-偶氮双[2-甲基-n-(2-羟基乙基)丙酰胺]、2,2-偶氮双(2,4,4-三甲基戊烷)、2-氰基-2-丙基偶氮甲酰胺、2,2-偶氮双(n-丁基-2-甲基丙酰胺)、2,2-偶氮双(n-环己基-2-甲基丙酰胺)等。
[0160]
第二工序中若使用预先包含聚合引发剂的状态的聚合性单体,则形成o/w型乳液时,在乳液的液滴中包含聚合引发剂,因此在后述的第三工序中,使乳液的液滴内部的单体聚合的聚合反应易于进行。
[0161]
第二工序中的聚合性单体与聚合引发剂的重量比并无特别限定,通常相对于聚合性单体100质量份、聚合引发剂优选为0.1质量份以上。聚合引发剂小于0.1质量份时,聚合反应不会充分地进行、复合粒子5的收量降低,因此不优选。
[0162]
另外,聚合性单体中还可以预先包含除聚合引发剂以外的其它功能性成分。作为具体例,可以示例磁性体、药品、农药、香料、粘接剂、酶、颜料、染料、除臭剂、金属、金属氧化物、无机氧化物等。聚合性单体中预先包含除聚合引发剂以外的其它功能性成分时,可以使所制得的复合粒子5的芯粒子内部含有功能性成分,可以表现出对应于用途的功能。
[0163]
(第三工序)
[0164]
第三工序是使包含芯粒子前体2的液滴固体化,如图3所示,获得芯粒子3被微细化纤维素层10被覆的复合粒子5的工序。
[0165]
将液滴进行固体化的具体方法根据液滴的材质而变化。使用聚合性单体时,进行聚合的方法并无特别限定,可以根据所用聚合性单体的种类及聚合引发剂的种类适当决定。作为聚合法的一例,可举出悬浮聚合法。
[0166]
具体的悬浮聚合的方法也无特别限定,可以使用公知的方法实施。
[0167]
例如,通过一边搅拌第二工序中获得的o/w型乳液一边加热,可以将液滴固体化。搅拌的方法也并无特别限定,可以使用公知的方法,具体地可以使用分散器或搅拌棒。
[0168]
还有不进行搅拌、仅通过加热处理即可固体化的情况。加热时的温度条件可以根据聚合性单体的种类及聚合引发剂的种类适当决定,例如可以为20℃以上且150℃以下。小于20℃时,有聚合的反应速度降低的情况,超过150℃时,有微细化纤维素1发生改性的情况。
[0169]
供至聚合反应的时间可以根据聚合性单体的种类及聚合引发剂的种类适当决定,例如可以为1小时~24小时左右。
[0170]
聚合反应可以通过作为电磁波一种的紫外线照射处理来实施。另外,除了电磁波以外,还可以使用电子束等粒子射线。
[0171]
使用热塑性聚合物时,通过使熔融了的聚合物发生相转移来使其固体化。作为相转移的方法,冷却是典型的。此时,通过控制冷却速度,可以控制热塑性聚合物的结晶化度。作为冷却的具体方法,可以示例使其扩散至水或冰水中的方法、使其接触于液氮等冷媒的方法、放冷的方法等。
[0172]
使用非固化性聚合物时,通过将溶剂除去来使其固体化。作为除去溶剂的方法并无特别限定,可以示例进行加热的方法或进行减压的方法、照射电磁波的方法、及它们的组合。
[0173]
当第三工序结束时,获得以聚合物为主成分的芯粒子3被微细化纤维素1被覆而成的大致圆球状的复合粒子5。在刚得到的复合粒子5中,表面存在的微细化纤维素1的至少一部分上结合有有机鎓阳离子/铵离子7a。另外,复合粒子5的粒径比较一致、均匀度高。
[0174]
第三工序刚刚结束后的分散液成为复合粒子5、大量的水、芯粒子3、未一体化而游离的微细化纤维素1混在的状态。
[0175]
作为从该分散液中仅取出复合粒子5时的回收、纯化方法,可以示例利用离心分离的洗涤、过滤洗涤等。作为利用离心分离的洗涤方法,可以使用公知的方法。例如,可以反复进行利用离心分离使分散液中的复合粒子5沉降后将上清除去、然后再分散于水-甲醇混合溶剂的操作,将残留溶剂从最终通过离心分离获得的沉降物中除去,从而将复合粒子5回收。对于过滤洗涤也是可以使用公知的方法。例如,使用可以孔径为0.1μm的ptfe滤膜,利用水和甲醇反复进行抽滤,从最终残留在滤膜上的糊状物中将残留溶剂除去,从而将复合粒子5回收。
[0176]
在任一情况下,残留溶剂的除去方法均无特别限定,可以通过风干或者通过利用烘箱热干燥来实施。包含复合粒子5的干燥固形物不会变为膜状或凝聚体状,而是以细腻的粉体的形式获得。
[0177]
(第四工序)
[0178]
第四工序是从所生成的复合粒子5中将有机鎓阳离子/铵离子7a除去的工序。第四工序根据需要在第三工序之后进行,鉴于复合粒子5的用途等如果不需要,则也可以省略。
[0179]
如上所述,在刚制造后的复合粒子5中,微细化纤维素1的一部分作为抗衡离子具有有机鎓阳离子/铵离子7a。关于复合粒子5的用途等,在不优选有机鎓阳离子/铵离子7a存在的情况下,或者想要将不同于有机鎓阳离子/铵离子7a的阳离子性物质以离子键的形式结合于微细化纤维素1的情况下,也可以进行第四工序来将有机鎓阳离子/铵离子7a除去。
[0180]
作为除去有机鎓阳离子/铵离子7a的方法,可以举出离子交换。在包含酸性化合物的水溶液中分散具有有机鎓阳离子/铵离子7a的复合粒子5,然后利用纯水进行洗涤,从而可以将有机鎓阳离子/铵离子7a除去。也可以在除去有机鎓阳离子/铵离子7a之后添加所希望的阳离子性化合物,在微细化纤维素1的阴离子性官能团上通过离子键结合不同于有机鎓阳离子/铵离子7a的阳离子性物质。
[0181]
如以上说明的那样,本实施方式的复合粒子5具有来自于在表面作为微细化纤维素层10存在的微细化纤维素1的高生物体亲和性及良好的分散稳定性。
[0182]
另外,由于使用结合有有机鎓阳离子/铵离子7a的微细化纤维素1形成,因此可以用宽范围种类的树脂来形成芯粒子3,获得能够对应于多种用途的复合粒子。例如,以往制造困难的具有热塑性的复合粒子也可以简单地制造。
[0183]
进而,微细化纤维素1通过有机鎓阳离子/铵离子7a而获得疏水性,变为双亲性。结果,复合粒子5的收率明显提高,同时粒径分布也均匀化,作为材料也变得优异。
[0184]
复合粒子5的干燥固形物在身为发挥微细化纤维素1的材料特性的物质的同时,还作为细腻的粉体获得,由于没有粒子之间的凝聚,因此也易于再次分散于溶剂中。由于微细化纤维素1与芯粒子3不可分地结合,因此在再分散后也会显示来自微细化纤维素1特性的稳定的分散。
[0185]
本实施方式的复合粒子5的制造方法可以在干燥状态下、以能够流通的状态简单地获得发挥微细化纤维素1的特性的粒子。因此,还可以期待对环境的负荷低、可削减输送费用、可减少腐败风险、提高作为添加剂的添加效率、提高在疏水性树脂中的混炼效率等效果。
[0186]
对于本发明的实施例,使用实施例进一步进行说明。本发明的技术范围并不受实施例的具体内容的任何限制。
[0187]
在以下的说明中,“%”只要无特别说明,则是指质量%。
[0188]
《实施例1》
[0189]
(第一工序:获得微细化纤维素分散液的工序)
[0190]
(木材纤维素的tempo氧化)
[0191]
将针叶树牛皮纸浆70g悬浮在蒸馏水3500g中,添加在蒸馏水350g中溶解有0.7g的tempo、7g的溴化钠的溶液,冷却至20℃。通过滴加向其中添加2mol/l、密度为1.15g/ml的次氯酸钠水溶液450g,引发氧化反应。体系内的温度一直保持在20℃,在反应中通过添加0.5n的氢氧化钠水溶液而将ph持续保持在10。在相对于纤维素的重量、氢氧化钠的总添加量达到3.50mmol/g的时刻,添加约100ml的乙醇,使反应停止。之后,使用玻璃过滤器反复进行利用蒸馏水的过滤洗涤,获得氧化纸浆。将所得氧化纸浆添加到调节为ph2的氯化氢水溶液中,脱水后进行水洗,进行第一次的酸处理。接着,添加到调节为ph3的氯化氢水溶液中,脱
水后进行水洗,进行第二次的酸处理。之后,反复进行水洗、脱水,获得纯化的氧化纸浆。
[0192]
(氧化纸浆的ir测定)
[0193]
在tempo氧化反应后,对于实施利用蒸馏水的水洗后的氧化纸浆(酸处理前)及之后利用氯化氢水溶液进行酸处理后实施水洗的氧化纸浆(酸处理后),用载玻片夹持,在70℃下干燥3小时,利用atr法实施ft-ir测定(日本分光公司制、ft/ir-6300)。将结果示于图4中。
[0194]
在酸处理后的氧化纸浆中,观察到来自cooh基的1720cm-1
附近的峰、但酸处理前观察到的来自coo-的1600cm-1
附近的峰消失。由以上确认了,通过酸处理,氧化纸浆的coo-基(na型)被完全置换成cooh基(h型)。
[0195]
(氧化纸浆的羧基量测定)
[0196]
称取以固体成分重量计为0.1g的tempo氧化后的氧化纸浆,以1%浓度将其分散在水中,添加盐酸,使ph为2.5。之后,利用使用了0.5m氢氧化钠水溶液的电导度滴定法计算羧基量(mmol/g)时,为1.6mmol/g。
[0197]
(通过抗衡离子置换来导入有机鎓阳离子)
[0198]
在氧化纸浆中添加水,制备固体成分浓度为5%的悬浮液,添加相对于氧化纸浆的羧基为1.0当量的作为碱种的有机鎓化合物即氢氧化四丁基铵(tbah)。使用搅拌器搅拌1小时,获得通过抗衡离子置换而导入了有机鎓阳离子的氧化纸浆。
[0199]
(抗衡离子置换氧化纸浆的解纤处理)
[0200]
将通过上述方法获得的抗衡离子置换氧化纸浆分散在蒸馏水中,利用榨汁机进行30分钟微细化处理。根据需要,利用离心脱泡器进行脱泡。由此,获得浓度为1%的微细化纤维素(tempo氧化cnf、csnf)分散液。所得分散液显示高的透明性和触变性。
[0201]
(第二工序:制作o/w型乳液的工序)
[0202]
作为芯粒子前体,使用作为聚合性单体的单官能性丙烯酸酯、丙烯酸异冰片酯(以下也称作“ib-xa”)10g,溶解1g作为聚合引发剂的2,2-偶氮双-2,4-二甲基戊腈(以下也称作“advn”)。向浓度为1%的微细化纤维素分散液40g添加ib-xa/advn混合溶液总量。ib-xa/advn混合溶液和分散液以各自透明性高的状态分离成2相。
[0203]
接着,从2相分离的状态的混合液中的上相的液面插入超声波匀浆机的旋转轴,在频率为24khz、输出功率为400w的条件下,进行3分钟的超声波匀浆机处理。超声波匀浆机处理后的混合液变成白浊的乳化液的状态。将一滴混合液滴加在载玻片上,用盖玻璃进行封入,利用光学显微镜进行观察时,观察到多个1~数μm左右的ib-xa的乳液液滴,确认到了以o/w型乳液的形式进行了分散稳定化的样子。
[0204]
(第三工序:通过芯粒子前体的固体化、获得被微细化纤维素被覆的复合粒子的工序)
[0205]
使用水浴槽将o/w型乳液分散液供至70℃的热水浴,一边利用搅拌棒搅拌一边处理8小时,实施聚合反应。在处理8小时后,将上述分散液冷却至室温,将液滴固体化,在分散液中生成复合粒子。在聚合反应的前后,分散液的外观没有变化。
[0206]
对所得的分散液进行离心分离(75000g、5分钟),获得包含复合粒子的沉降物。通过倾析将上清除去,将沉降物回收,进而使用孔径为0.1μm的ptfe滤膜,利用纯水和甲醇反复洗涤。将如此经纯化、回收的复合粒子以1%浓度再分散于纯水中,使用粒度分布计
(nanotrac upa-ex150、日机装株式会公司制)测定粒径时,平均粒径(中位值)为2.5μm。对复合粒子进行风干,在室温25度下实施24小时的真空干燥处理时,变为细腻的干燥粉体,未发生凝聚或膜状化。
[0207]
(利用sem的复合粒子的形状观察)
[0208]
将上述干燥粉体的sem图像示于图5及图6中。图5的倍率为100倍,图6的倍率在左上区域中为2000倍、其它区域中为50000倍。第二工序及第三工序中,通过以o/w型乳液液滴为模板实施聚合反应,获得多个来自于乳液液滴的形状的圆球状的复合粒子5,由图5可知,直径的均匀度也高。
[0209]
由图6可知,复合粒子的表面被宽度为数nm左右的微细化纤维素1均匀地被覆。图6由于是反复过滤洗涤后的图像,因此表示在本发明的复合粒子5中,芯粒子3与微细化纤维素1结合在一起、处于不可分的状态。
[0210]
(复合粒子的粒度分布)
[0211]
将利用beckman coulter公司制的粒度分布计ls-13320测定上述干燥粉体的粒度分布的结果示于图9中。粒度分布以2.5μm为中央值、基本集中在1~10μm的范围中,显示与sem图像良好的一致。
[0212]
《实施例2~4》
[0213]
除了代替tbah使用以下的有机鎓化合物之外,利用与实施例1相同的顺序制作实施例2~4的复合粒子。
[0214]
实施例2氢氧化四甲基铵(tmah)
[0215]
实施例3二甲基苄基胺(dmba)
[0216]
实施例4二甲基硬脂基胺(dmsa)
[0217]
《实施例5》
[0218]
除了代替ib-xa使作为单官能性甲基丙烯酸酯的甲基丙烯酸异冰片酯(ib-x)作为芯粒子前体之外,利用与实施例1相同的顺序制作实施例5的复合粒子。
[0219]
《实施例6》
[0220]
除了代替ib-xa使作为单官能性乙烯基单体的对甲基苯乙烯(p-mest)作为芯粒子前体之外,利用与实施例1相同的顺序制作实施例6的复合粒子。
[0221]
《实施例7》
[0222]
除了代替tempo氧化cnf使用专利文献2记载的进行羧甲基化(以下也称作“cm化”)处理而获得的cm化cnf之外,在与实施例1相同的条件下制作实施例7的复合粒子。cm化cnf显示高透明性。
[0223]
《实施例8》
[0224]
作为芯粒子前体,使用聚己内酯(pcl、分子量为10,000)。在第二工序中,将pcl的20%mek溶液添加在微细化纤维素分散液中。将该分散液加热至75℃,进行超声波匀浆机处理,制成o/w型乳液之后,用冰水进行冷却,将液滴固体化。
[0225]
除此之外,在与实施例1相同的条件下制作实施例7的复合粒子。
[0226]
《实施例9》
[0227]
除了代替tbah使用dmba之外,利用与实施例8相同的顺序制作实施例9的复合粒子。
[0228]
《实施例10》
[0229]
除了使tbah的添加量相对于氧化纸浆的羧基为0.8当量之外,利用与实施例1相同的顺序制作实施例10的复合粒子。
[0230]
《实施例11》
[0231]
除了使tbah的添加量相对于氧化纸浆的羧基为1.8当量之外,利用与实施例1相同的顺序制作实施例11的复合粒子。
[0232]
《实施例12》
[0233]
除了在第一工序中使酸处理仅为1次之外,利用与实施例1相同的顺序制作实施例12的复合粒子。
[0234]
实施例12中,利用ft-ir测定求得的氧化纸浆的cooh基的置换度为0.7。置换度的定义是:以ch伸缩基来源的2870cm-1
的峰强度为基准,计算1720cm-1
的峰强度比(i1720/i2870),将使图5中的i1720/i2870为1时的比作为置换度。
[0235]
《实施例13、14》
[0236]
除了代替有机鎓化合物tbah使用以下的胺之外,利用与实施例1相同的顺序制作实施例13~14的复合粒子。
[0237]
实施例13硬脂基胺
[0238]
实施例14三己基胺
[0239]
《比较例1》
[0240]
除了使用未进行tempo氧化处理的未处理纸浆之外,利用与实施例1相同的顺序制作比较例1的复合粒子。
[0241]
《比较例2》
[0242]
除了代替tbah使用并非有机鎓化合物的氢氧化钠之外,利用与实施例1相同的顺序制作比较例2的复合粒子。
[0243]
《比较例3》
[0244]
除了代替tbah使用并非有机鎓化合物的氢氧化钠之外,利用与实施例5相同的顺序制作比较例3的复合粒子。
[0245]
《比较例4》
[0246]
除了代替tbah使用并非有机鎓化合物的氢氧化钠之外,利用与实施例6相同的顺序制作比较例4的复合粒子。
[0247]
《比较例5》
[0248]
除了代替tbah使用并非有机鎓化合物的氢氧化钠之外,利用与实施例7相同的顺序制作比较例5的复合粒子。
[0249]
《比较例6》
[0250]
除了代替tbah使用并非有机鎓化合物的氢氧化钠之外,利用与实施例8相同的顺序制作比较例6的复合粒子。
[0251]
(第四工序:将有机鎓阳离子从复合粒子5中除去的工序)
[0252]
将实施例1的复合粒子5的干燥粉体按照固体成分浓度达到1%的方式添加在ph2.5的氯化氢水溶液中,利用超声波洗涤机处理5分钟,进而使用搅拌器搅拌30分钟。由此获得目视下没有凝聚的悬浮液。使用离心分离(25000g、10分钟)及倾析将该悬浮液浓缩,接
着添加在ph4的氯化氢水溶液中,搅拌器搅拌5分钟之后,利用离心分离及倾析进一步进行浓缩。之后,利用纯水反复5次的洗涤和浓缩。对纯化、回收的复合粒子进行风干,进而在室温25度下实施24小时的真空干燥处理,从而获得干燥粉体。对所得干燥粉体进行xps解析时,未检测到来自于tbah的氮源,确认到除去了有机鎓阳离子。再次将干燥粉体添加到纯水中,进行超声波处理时,显示了良好的再分散。
[0253]
对所有的实施例进行上述处理,确认到了有机鎓阳离子的除去及良好的再分散。对于比较例,未进行第四工序。
[0254]
表1中一并示出实施例及比较例的内容。
[0255]
表1
[0256][0257]
表2表示实施例及比较例的评价结果。评价项目及标准如下所述。
[0258]
·
复合粒子的收率(%)
[0259]
以所获取的复合粒子的重量(g)/制造中使用了的芯粒子前体的树脂重量(g)
×
100算出。
[0260]
·
复合粒子的平均粒径(中位值):使用激光衍射式粒度分布计(beckman coulter公司制、ls-13320)求得。当存在粗大化的树脂的块时,将其除去后进行测定。
[0261]
·
各工序可否执行
[0262]
(第一工序)
[0263]
ο(可以执行):对于分散液,利用分光光度计测定的透射率(光路长为1cm、波长为600nm)为70%以上
[0264]
×
(不可以执行):对于分散液,利用分光光度计测定的透射率(光路长为1cm、波长为600nm)小于70%
[0265]
分散液的透射率低表示纤维素未发生微细化。
[0266]
(第二工序)
[0267]
ο(可以执行):24小时静止后,o/w型乳液的总光线透射率未见变化。
[0268]
×
(不可以执行):24小时静置后,o/w型乳液的总光线透射率发生大的变化。或可见分离。
[0269]
乳液的总光线透射率的变化是由于乳液的分离或合一等不稳定化而产生的。
[0270]
(第三工序)
[0271]
ο(可以执行):获得了多个球状的复合粒子
[0272]
×
(不可以执行):可见粒子并非球状、或复合粒子凝聚而成的粗大的块
[0273]
乳液的稳定性高时,在聚合后也会维持乳液的液滴的形状。而在聚合前或聚合中乳液发生不稳定化时,由于分离或合一,会产生异形的树脂或粗大化的树脂。
[0274]
表2
[0275][0276]
如表2所示,在各实施例中,在微细化纤维素分散液中,芯粒子前体稳定地进行液滴化,良好地以高收率形成了粒径小且均匀的复合粒子。认为其原因在于,通过有机鎓化合物来源的有机鎓阳离子,亲水性的cnf的一部分被疏水化,对芯粒子前体的吸附力有所提高。
[0277]
在任一个比较例中均无法良好地进行第一工序~第三工序的全部。结果,复合粒子的收率也低、粒径的均匀度也低。
[0278]
图7及图8表示利用sem观察比较例1的复合粒子的干燥粉体的照片。比较例1中,由图7可知,存在粗大化的复合粒子、粒度分布不均匀。在粗大化的复合粒子中,如图8所示,芯粒子表面的大部分露出,微细化纤维素仅稍有结合。
[0279]
产业上的可利用性
[0280]
本发明的复合粒子从作为添加剂的添加效率、与树脂的混炼效率提高、以及输送效率提高或防止腐败的观点出发,从还有助于削减成本等产业实施的观点出发,可获得优选的效果。本复合粒子通过发挥构成粒子表面的微细化纤维素及构成芯粒子的聚合物的特性,可以适用于色料、吸附剂、化妆颜料、缓释材料、除臭剂、抗菌性医疗用构件、面向个人护理用品的抗菌性物品、包装材料、色素增敏型太阳能电池、光电转换材料、光热转换材料、隔
热材料、光学滤波器、拉曼增强元件、图像显示元件、磁性粉、催化剂担载体、药物传递系统等。
[0281]
符号说明
[0282]
1 微细化纤维素
[0283]
2 芯粒子前体(液滴)
[0284]
3 芯粒子
[0285]
5 复合粒子
[0286]
6 纤维素原料
[0287]
7 有机鎓化合物/胺
[0288]
7a 有机鎓阳离子/铵离子
[0289]
10 微细化纤维素层