1.本发明属于药物合成和材料化学技术领域,具体涉及一种新型三嗪类衍生物及合成方法。
背景技术:
2.1,3,5-三嗪类衍生物由于具有较好的载流子传输能力,其作为电子传输材料使用而制成的有机电致发光器件,表现出驱动电压低、发光效率高等优点,是性能优良的有机发光材料,从而广泛的存在于材料科学中。目前,文献中所报道的合成方法多为常见的suzuki偶联,锂试剂和格氏试剂法,其中suzuki偶联通常需要较高的能量去引发,且易发生热集聚导致剧烈放热,有冲料风险,而锂试剂和格氏试剂法反应活性非常高,这三类反应普遍存在反应条件苛刻,制备过程中易生成多取代的杂质,且难以除去。
技术实现要素:
3.本发明的目的在于克服上述现有技术的不足,提供了一种新型三嗪类衍生物及合成方法。
4.为了解决技术问题,本发明的技术方案是:一种新型三嗪类衍生物的合成方法,以芳香类卤代物和带有一个活性氯的1,3,5-三嗪类化合物为原料,在催化剂的催化下,得到1,3,5活性位均被芳香基替代的三嗪类衍生物,其中芳香类卤代物和带有一个活性氯的1,3,5-三嗪类化合物的摩尔比为1~1.5:1,催化剂的用量为带有一个活性氯的1,3,5-三嗪类化合物摩尔数的1%~3%。
5.优选的,所述步骤1具体为:将芳香类卤代物溶解于四氢呋喃并加入三口瓶中,开启搅拌,在惰性气体保护下将体系降温至-80℃~-90℃,将正丁基锂滴加至体系中并搅拌1~2h,接着将氯化锌溶解于四氢呋喃中,控温在-60℃~-80℃,将其滴加至体系中,然后将体系升温至25~30℃并搅拌1~1.5h,接着将体系降温至18~20℃,将带有一个活性氯的1,3,5-三嗪类化合物和催化剂加入体系中,使用惰性气体置换体系中的空气三次,将体系升温至50℃~55℃,并搅拌16~24h待反应完全,停止反应得到1,3,5活性位均被芳香基替代的三嗪类衍生物的反应体系,所述芳香类卤代物与四氢呋喃的用量比为1g:5~7ml,芳香类卤代物与正丁基锂的摩尔比为1:1~1.2,芳香类卤代物与氯化锌的摩尔比为1:1~1.2,氯化锌与四氢呋喃的用量比为1g:4~7ml。
6.优选的,所述芳香类卤代物和带有一个活性氯的1,3,5-三嗪类化合物的摩尔比为1.2:1,芳香类卤代物与四氢呋喃的用量比为1g:6ml,芳香类卤代物与正丁基锂的摩尔比为1:1,芳香类卤代物与氯化锌的摩尔比为1:1.1,氯化锌与四氢呋喃的用量比为1g:5ml。
7.优选的,所述惰性气体为氩气。
8.优选的,所述催化剂为四三苯基膦钯或1,1
’‑
二(二苯基膦)二茂铁二氯化钯,催化剂的用量为带有一个活性氯的1,3,5-三嗪类化合物摩尔数的2%。
9.优选的,所述体系搅拌16~24h待反应完全后取样检测原料,当原料剩余小于5%,
停止反应。
10.优选的,所述1,3,5活性位均被芳香基替代的三嗪类衍生物的反应体系后处理为:将1,3,5活性位均被芳香基替代的三嗪类衍生物的反应体系倒入乙酸乙酯、浓盐酸和水的混合体系中,充分搅拌萃取,分液弃去水相,有机相干燥后浓缩干得到固体粗品,使用乙酸乙酯重结晶一次,得到1,3,5活性位均被芳香基替代的三嗪类衍生物。
11.优选的,所述混合体系中的乙酸乙酯、浓盐酸和水的用量比为5:1:10,芳香类卤代物与混合体系的用量比为1g:16~19。
12.优选的,所述芳香类卤代物为间氟溴苯、间氯溴苯、间溴碘苯、对氟溴苯、对氯溴苯或对溴碘苯,所述带有一个活性氯的1,3,5-三嗪类化合物的结构式为:
13.其中r1、r2基团分别为苯基或联苯基。
14.优选的,一种新型三嗪类衍生物,由上述任一项所述的一种新型三嗪类衍生物的合成方法合成,新型三嗪类衍生物为1,3,5活性位均被芳香基替代的三嗪类衍生物,具体为:
15.2-(3-氟苯基)-4,6-二苯基-1,3,5-三嗪;
16.2-(3-氯苯基)-4,6-二苯基-1,3,5-三嗪;
17.2-(3-溴苯基)-4,6-二苯基-1,3,5-三嗪;
18.2-(4-氟苯基)-4,6-二苯基-1,3,5-三嗪;
19.2-(4-氯苯基)-4,6-二苯基-1,3,5-三嗪;
20.2-(4-溴苯基)-4,6-二苯基-1,3,5-三嗪;
21.2-(联苯-4-基)-4-(3-氟苯基)-6-苯基-1,3,5-三嗪;
22.2-(联苯-4-基)-4-(3-氯苯基)-6-苯基-1,3,5-三嗪;
23.2-(联苯-4-基)-4-(3-溴苯基)-6-苯基-1,3,5-三嗪;
24.2-(1,1'联苯-4-基)-4-(4-氟苯基)-6-苯基-1,3,5-三嗪;
25.2-(1,1'联苯-4-基)-4-(4-氯苯基)-6-苯基-1,3,5-三嗪;
26.2-(1,1'联苯-4-基)-4-(4-溴苯基)-6-苯基-1,3,5-三嗪。
27.与现有技术相比,本发明的优点在于:
28.(1)本发明研发了一种温和的制备三嗪类衍生物的方法,以多种芳香类卤代物的锌试剂和一类带有一个活性氯的1,3,5-三嗪类化合物为原料,利用锌试剂来做金属迁移,在四三苯基膦钯或者1,1
’‑
二(二苯基膦)二茂铁二氯化钯催化下,得到一系列的1,3,5活性位均被芳香基替代的三嗪类衍生物;本发明三嗪类衍生物是一类重要的药物活性和光电性能分子,在药物学和材料化学中有广泛的用途,使用有机锌试剂法制备三嗪类衍生物,产率达70%~90%;
29.(2)本发明反应条件温和,可以让反应平顺的进行,这样可以保证大规模生产,相较于传统的suzuki偶联法,本发明方法选择性好,能较好地控制多取代杂质,且易于提纯;
30.(3)本发明中使用的有机锌试剂相较于其他文献中的锂试剂和格氏试剂稳定,且
制备简单,添加主料可以在常温环境中进行,整个反应过程温和,不会有安全风险,无需锂试剂必须的超低温环境,也无格氏试剂制备过程中的高温放热冲料风险;
31.(4)本发明中所使用的的催化剂相较于其他催化剂制备简单,且便宜易得。
附图说明
32.图1、本发明实施例1中2-(3-氟苯基)-4,6-二苯基-1,3,5-三嗪的1h-nmr谱图;
33.图2、本发明实施例1中2-(3-氟苯基)-4,6-二苯基-1,3,5-三嗪的
13
c-nmr谱图;
34.图3、本发明实施例2中2-(联苯-4-基)-4-(3-溴苯基)-6-苯基-1,3,5-三嗪的1h-nmr谱图;
35.图4、本发明实施例2中2-(联苯-4-基)-4-(3-溴苯基)-6-苯基-1,3,5-三嗪的
13
c-nmr谱图;
36.图5、本发明实施例3中2-(1,1'联苯-4-基)-4-(4-氯苯基)-6-苯基-1,3,5-三嗪的1h-nmr谱图;
37.图6、本发明实施例3中2-(1,1'联苯-4-基)-4-(4-氯苯基)-6-苯基-1,3,5-三嗪的
13
c-nmr谱图。
具体实施方式
38.以下结合具体实施例对本发明进行说明,所用的原材料、溶剂和催化剂均为常规市售产品,以下实施例用于说明本发明,但不用来限制本发明的范围。
39.本发明公开了一种新型三嗪类衍生物的合成方法,以芳香类卤代物和带有一个活性氯的1,3,5-三嗪类化合物为原料,在催化剂的催化下,得到1,3,5活性位均被芳香基替代的三嗪类衍生物,其中芳香类卤代物和带有一个活性氯的1,3,5-三嗪类化合物的摩尔比为1~1.5:1,催化剂的用量为带有一个活性氯的1,3,5-三嗪类化合物摩尔数的1%~3%。
40.优选的,所述步骤1具体为:将芳香类卤代物溶解于四氢呋喃并加入三口瓶中,开启搅拌,在惰性气体保护下将体系降温至-80℃~-90℃,将正丁基锂滴加至体系中并搅拌1~2h,接着将氯化锌溶解于四氢呋喃中,控温在-60℃~-80℃,将其滴加至体系中,然后将体系升温至25~30℃并搅拌1~1.5h,接着将体系降温至18~20℃,将带有一个活性氯的1,3,5-三嗪类化合物和催化剂加入体系中,使用惰性气体置换体系中的空气三次,将体系升温至50℃~55℃,并搅拌16~24h待反应完全,停止反应得到1,3,5活性位均被芳香基替代的三嗪类衍生物的反应体系,所述芳香类卤代物与四氢呋喃的用量比为1g:5~7ml,芳香类卤代物与正丁基锂的摩尔比为1:1~1.2,芳香类卤代物与氯化锌的摩尔比为1:1~1.2,氯化锌与四氢呋喃的用量比为1g:4~7ml。
41.优选的,所述芳香类卤代物和带有一个活性氯的1,3,5-三嗪类化合物的摩尔比为1.2:1,芳香类卤代物与四氢呋喃的用量比为1g:6ml,芳香类卤代物与正丁基锂的摩尔比为1:1,芳香类卤代物与氯化锌的摩尔比为1:1.1,氯化锌与四氢呋喃的用量比为1g:5ml。
42.优选的,所述惰性气体为氩气。
43.优选的,所述催化剂为四三苯基膦钯或1,1
’‑
二(二苯基膦)二茂铁二氯化钯,催化剂的用量为带有一个活性氯的1,3,5-三嗪类化合物摩尔数的2%。
44.优选的,所述体系搅拌16~24h待反应完全后取样检测原料,当原料剩余小于5%,
停止反应。
45.优选的,所述1,3,5活性位均被芳香基替代的三嗪类衍生物的反应体系后处理为:将1,3,5活性位均被芳香基替代的三嗪类衍生物的反应体系倒入乙酸乙酯、浓盐酸和水的混合体系中,充分搅拌萃取,分液弃去水相,有机相干燥后浓缩干得到固体粗品,使用乙酸乙酯重结晶一次,得到1,3,5活性位均被芳香基替代的三嗪类衍生物。
46.优选的,所述混合体系中的乙酸乙酯、浓盐酸和水的用量比为5:1:10,芳香类卤代物与混合体系的用量比为1g:16~19。
47.优选的,所述芳香类卤代物为间氟溴苯、间氯溴苯、间溴碘苯、对氟溴苯、对氯溴苯或对溴碘苯,所述带有一个活性氯的1,3,5-三嗪类化合物的结构式为:
48.其中r1、r2基团分别为苯基或联苯基。
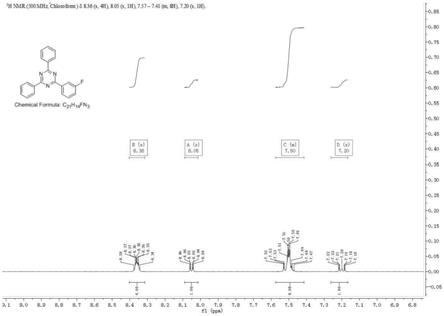
49.所述原料1,3,5-三嗪类衍生物r1、r2基团可以是苯基或联苯基等,可以顺利反应得到相应的三嗪类衍生物。
50.优选的,一种新型三嗪类衍生物,由上述任一项所述的一种新型三嗪类衍生物的合成方法合成,新型三嗪类衍生物为1,3,5活性位均被芳香基替代的三嗪类衍生物,具体为:
51.2-(3-氟苯基)-4,6-二苯基-1,3,5-三嗪;
52.2-(3-氯苯基)-4,6-二苯基-1,3,5-三嗪;
53.2-(3-溴苯基)-4,6-二苯基-1,3,5-三嗪;
54.2-(4-氟苯基)-4,6-二苯基-1,3,5-三嗪;
55.2-(4-氯苯基)-4,6-二苯基-1,3,5-三嗪;
56.2-(4-溴苯基)-4,6-二苯基-1,3,5-三嗪;
57.2-(联苯-4-基)-4-(3-氟苯基)-6-苯基-1,3,5-三嗪;
58.2-(联苯-4-基)-4-(3-氯苯基)-6-苯基-1,3,5-三嗪;
59.2-(联苯-4-基)-4-(3-溴苯基)-6-苯基-1,3,5-三嗪;
60.2-(1,1'联苯-4-基)-4-(4-氟苯基)-6-苯基-1,3,5-三嗪;
61.2-(1,1'联苯-4-基)-4-(4-氯苯基)-6-苯基-1,3,5-三嗪;
62.2-(1,1'联苯-4-基)-4-(4-溴苯基)-6-苯基-1,3,5-三嗪。
63.实施例1:
64.制备化合物1a(2-(3-氟苯基)-4,6-二苯基-1,3,5-三嗪):称取间氟溴苯42g(0.24mol)溶解于300ml四氢呋喃中,加入2l三口瓶中,开启搅拌,氩气保护下将体系降温至-80℃~-90℃以下,控温在-80℃~-90℃,将正丁基锂120ml(0.24mol)滴加至体系中,-80℃~-90℃搅拌1h,将氯化锌35.4g(0.26mol)溶解于150ml四氢呋喃中(溶解完是乳浊液,不是完全溶清状态,不影响滴加),控温在-60℃~-80℃,将其滴加至体系中,然后将体系升温至30℃,搅拌1.5h,将体系降温至20℃以下,将2-氯-4,6-二苯基-1,3,5-三嗪53.5g(0.2mol)、1,1
’‑
二(二苯基膦)二茂铁二氯化钯1.46g(2mmol)加入体系中,使用氩气置换体
系中的空气三次,将体系升温至55℃,50℃~55℃搅拌24h后取样检测,原料反应完全,停止反应。
65.反应完将体系倒入250ml乙酸乙酯、50ml浓盐酸和500ml水的混合体系中,充分搅拌萃取,分液弃去水相,有机相干燥后浓缩干得到固体粗品,使用乙酸乙酯150ml重结晶一次,得到58.2g白色固体,收率89%。
66.将合成的化合物采用1h-nmr和
13
c-nmr表征,谱图数据与结构吻合。
67.图1是由本技术合成方法合成的产物2-(3-氟苯基)-4,6-二苯基-1,3,5-三嗪的1h-nmr谱图,图中产物的氢原子在7.20ppm,7.41ppm-7.57ppm,8.05ppm,8.36ppm处有化学位移,且峰面积比为1:8:1:4。
68.图2是由本技术合成方法合成的产物2-(3-氟苯基)-4,6-二苯基-1,3,5-三嗪的
13
c-nmr谱图,图中峰值分别在115.19ppm,115.35ppm,117.91ppm,118.07ppm,126.41ppm,126.43ppm,128.46ppm,128.68ppm,130.20ppm,130.26ppm,131.69ppm,136.41ppm,138.32ppm,138.38ppm,162.17ppm,164.19ppm,167.35ppm,167.37ppm,171.35ppm。
69.实施例2
70.制备化合物3c(2-(联苯-4-基)-4-(3-溴苯基)-6-苯基-1,3,5-三嗪):称取间溴碘苯28.4g(0.1mol)溶解于170ml四氢呋喃中,加入1l三口瓶中,开启搅拌,氩气保护下将体系降温至-80℃~-90℃以下,控温在-80℃~-90℃,将正丁基锂60ml(0.12mol)滴加至体系中,-80℃~-90℃搅拌1h,将氯化锌17.7g(0.13mol)溶解于90ml四氢呋喃中,控温在-60℃~-80℃,将其滴加至体系中,然后将体系升温至30℃,搅拌2h,将体系降温至20℃以下,将2-(苯基-4-基)-4-氯-6-苯基-1,3,5-三嗪34.4g(0.1mol)、1,1
’‑
二(二苯基膦)二茂铁二氯化钯0.73g(2mmol)加入体系中,使用氩气置换体系中的空气三次,将体系升温至25℃,体系开始放热,适当用冷水浴降温,控制体系温度不超过35℃,25℃~35℃搅拌16h后取样检测,原料剩余小于5%,二取代杂质小于5%,停止反应。
71.反应完将体系倒入170ml乙酸乙酯、35ml浓盐酸和350ml水的混合体系中,充分搅拌萃取,分液弃去水相,有机相干燥后浓缩干得到固体粗品,使用二氯乙烷100ml和乙酸乙酯170ml混合体系重结晶一次,得到33.4g白色固体,收率72%。
72.将合成的化合物采用1h-nmr和
13
c-nmr表征,谱图数据与结构吻合。
73.图3是由本技术合成方法合成的产物2-(联苯-4-基)-4-(3-溴苯基)-6-苯基-1,3,5-三嗪的1h-nmr谱图,图中产物的氢原子在7.21ppm-7.29ppm,7.35ppm-7.54ppm,7.58ppm-7.66ppm,7.71ppm-7.79ppm,7.92ppm-7.99ppm,8.22ppm,8.31ppm-8.40ppm处有化学位移,且峰面积比为2:8:1:2:2:1:2
74.图4是由本技术合成方法合成的产物2-(联苯-4-基)-4-(3-溴苯基)-6-苯基-1,3,5-三嗪的
13
c-nmr谱图,图中峰值分别在123.41ppm,127.00ppm,127.56ppm,127.72ppm,128.46ppm,128.59ppm,128.68ppm,128.87ppm,130.44ppm,130.86ppm,131.69ppm,132.54ppm,135.11ppm,136.41ppm,137.21ppm,138.60ppm,139.23ppm,146.20ppm,167.36ppm,170.71ppm,171.35ppm。
75.实施例3
76.制备化合物4b(2-(1,1'联苯-4-基)-4-(4-氯苯基)-6-苯基-1,3,5-三嗪):称取对氯溴苯41.6g(217.8mmol)溶解于165ml四氢呋喃中,加入1l三口瓶中,开启搅拌,氩气保护
下将体系降温至-80℃~-90℃以下,控温在-80℃~-90℃,将正丁基锂87ml(174.5mmol)滴加至体系中,-80℃~-90℃搅拌0.5h,将氯化锌25.8g(189mmol)溶解于125ml四氢呋喃中,控温在-60℃~-80℃,将其滴加至体系中,然后将体系升温至30℃,搅拌2h,将体系降温至20℃以下,将2-(苯基-4-基)-4-氯-6-苯基-1,3,5-三嗪50g(145.4mmol)、四三苯基膦钯1.67g(1.45mmol)加入体系中,使用氩气置换体系中的空气三次,将体系升温至25℃,体系开始放热,适当用冷水浴降温,控制体系温度不超过30℃,25℃~30℃搅拌20h后取样检测,原料剩余小于3%,二取代杂质小于5%,停止反应。
77.反应完将体系倒入250ml乙酸乙酯、50ml浓盐酸和500ml水的混合体系中,充分搅拌萃取,分液弃去水相,有机相干燥后浓缩干得到固体粗品,使用乙酸乙酯150ml回流煮洗一次,得到48.9g白色固体,收率80%。
78.将合成的化合物采用1h-nmr和
13
c-nmr表征,谱图数据与结构吻合。
79.图5是由本技术合成方法合成的产物2-(1,1'联苯-4-基)-4-(4-氯苯基)-6-苯基-1,3,5-三嗪的1h-nmr谱图,图中产物的氢原子在7.21ppm-7.30ppm,7.36ppm-7.55ppm,7.73ppm-7.78ppm,7.91ppm-8.00ppm,8.21ppm-8.29ppm,8.31ppm-8.42ppm处有化学位移,且峰面积比为2:8:2:2:2:2。
80.图6是是由本技术合成方法合成的产物2-(1,1'联苯-4-基)-4-(4-氯苯基)-6-苯基-1,3,5-三嗪的
13
c-nmr谱图,图中峰值分别在130.35ppm,130.45ppm,131.55ppm,131.69ppm,135.36ppm,136.41ppm,136.54ppm,137.21ppm,139.23ppm,146.20ppm,170.76ppm,170.86ppm,171.79ppm。
81.本发明其他三嗪类衍生物制备过程为:将芳香类卤代物溶解于四氢呋喃并加入三口瓶中,开启搅拌,在惰性气体保护下将体系降温至-80℃~-90℃,将正丁基锂滴加至体系中并搅拌1~2h,接着将氯化锌溶解于四氢呋喃中,控温在-60℃~-80℃,将其滴加至体系中,然后将体系升温至25~30℃并搅拌1~1.5h,接着将体系降温至18~20℃,将带有一个活性氯的1,3,5-三嗪类化合物和催化剂加入体系中,使用惰性气体置换体系中的空气三次,将体系升温至50℃~55℃,并搅拌16~24h待反应完全,停止反应得到1,3,5活性位均被芳香基替代的三嗪类衍生物的反应体系,芳香类卤代物和带有一个活性氯的1,3,5-三嗪类化合物的摩尔比为1~1.5:1,催化剂的用量为带有一个活性氯的1,3,5-三嗪类化合物摩尔数的1%~3%,所述芳香类卤代物与四氢呋喃的用量比为1g:5~7ml,芳香类卤代物与正丁基锂的摩尔比为1:1~1.2,芳香类卤代物与氯化锌的摩尔比为1:1~1.2,氯化锌与四氢呋喃的用量比为1g:4~7ml。
82.本发明其他三嗪类衍生物的收率如下所示:
[0083][0084]
本发明反应原理如下:
[0085][0086]
本发明以多种芳香类卤代物的锌试剂和一类带有一个活性氯的1,3,5-三嗪类化合物为原料,在四三苯基膦钯或者1,1
’‑
二(二苯基膦)二茂铁二氯化钯催化下,25~55℃搅拌下反应,得到一系列的1,3,5活性位均被芳香基替代的三嗪类衍生物。
[0087]
本发明反应原理与其他偶联反应原理基本一致,同样要经过氧化加成,金属转移和还原消除三个阶段,流程如下:
[0088][0089]
本发明研发了一种温和的制备三嗪类衍生物的方法,以多种芳香类卤代物的锌试剂和一类带有一个活性氯的1,3,5-三嗪类化合物为原料,利用锌试剂来做金属迁移,在四三苯基膦钯或者1,1
’‑
二(二苯基膦)二茂铁二氯化钯催化下,得到一系列的1,3,5活性位均被芳香基替代的三嗪类衍生物;本发明三嗪类衍生物是一类重要的药物活性和光电性能分子,在药物学和材料化学中有广泛的用途,使用有机锌试剂法制备三嗪类衍生物,产率达
70%~90%。
[0090]
本发明反应条件温和,可以让反应平顺的进行,这样可以保证大规模生产,相较于传统的suzuki偶联法,本发明方法选择性好,能较好地控制多取代杂质,且易于提纯。
[0091]
本发明中使用的有机锌试剂相较于其他文献中的锂试剂和格氏试剂稳定,且制备简单,添加主料可以在常温环境中进行,整个反应过程温和,不会有安全风险,无需锂试剂必须的超低温环境,也无格氏试剂制备过程中的高温放热冲料风险;本发明中所使用的的催化剂相较于其他催化剂制备简单,且便宜易得。
[0092]
上面对本发明优选实施方式作了详细说明,但是本发明不限于上述实施方式,在本领域普通技术人员所具备的知识范围内,还可以在不脱离本发明宗旨的前提下做出各种变化。
[0093]
不脱离本发明的构思和范围可以做出许多其他改变和改型。应当理解,本发明不限于特定的实施方式,本发明的范围由所附权利要求限定。