1.本发明属于药物领域,涉及一种吉西他滨环磷酸酯前药及其制备方法和应用。
背景技术:
2.核苷类药物在肿瘤、感染性疾病等领域应用广泛,其作用靶点多为dna聚合酶、rna聚合酶、rna逆转录酶。核苷类药物一般模拟天然核苷的结构,竞争性地作用于酶活性中心,嵌入正在合成的dna或rna链中,干扰核酸代谢。核苷类药物进入细胞后在酶的作用下经三步磷酸化,得到具有生物活性的三磷酸衍生物而发挥药效,其中单磷酸化是限速步骤,因此通常在核苷类药物中直接引入单磷酸或磷酸酯基团。但早期设计的磷酸或磷酸酯衍生物因其极性大难以通过细胞膜,磷-氧键代谢稳定性较差等限制了应用。因此,为了解决类药性问题,在核苷类药物的设计中,前药策略被广泛采用。
3.protide前药技术的设计原理是将核苷磷酸/磷酸类药物分别通过磷酯键/磷酰胺键(芳基模块/氨基酸酯基模块)与极性基团连接形成磷酯/磷酰胺前药,通过掩蔽极性基团来降低分子极性增加透膜性,当前药吸收进入体内后再经特定酶水解释放原型药物(acs med.chem.lett.2019,10,2-5.)。fda已批准三个protide前药:用于治疗乙肝的替诺福韦艾拉酚胺(tenefovir alafenamide,taf)、治疗慢性丙肝的索非布韦(sofosbuvir)和治疗新冠肺炎的瑞德西韦(remdesivir)。
4.吉西他滨是一种嘧啶类抗肿瘤药物,其主要代谢物在细胞内掺入dna并作用于g1/s期,其还能通过抑制核苷酸还原酶导致细胞内脱氧核苷三磷酸酯的减少(br j cancer.1996,73,101.)。但核苷类抗肿瘤药物容易出现耐药性,其protide前药能够减少耐药发生,具有良好的抗肿瘤作用(j.med.chem.2014,57,1531-1542.)。
技术实现要素:
5.本发明提供了一种吉西他滨环磷酸酯前药及其制备方法和应用。通过药理学实验证明,本发明得到的吉西他滨环磷酸酯前药具有显著的抗肿瘤作用。
6.为实现上述发明目的,本发明的技术方案如下:
7.本发明提供了一种吉西他滨环磷酸酯前药,或其光学异构体、非对映异构体、消旋体或三者的混合物,或其药学上可接受的盐、溶剂合物、氘代物,具有如下通式的结构:
[0008][0009]
其中:
[0010]
r选自氢、甲基、乙基、丙基、异丙基、正丁基、异丁基、叔丁基、苄基或苯环取代的苄基。
[0011]
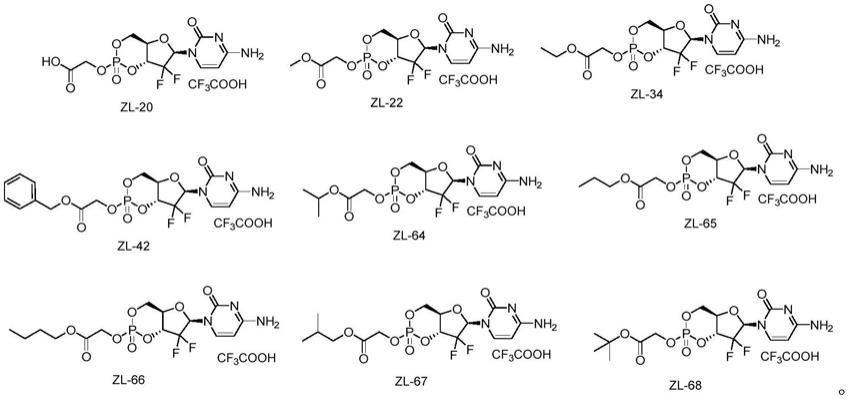
进一步的:所述吉西他滨环磷酸酯前药具体为zl-20、zl-22、zl-34、zl-42、zl-64、zl-65、zl-66、zl-67、zl-68,其结构式分别如下:
[0012][0013]
本发明还提供了所述的吉西他滨环磷酸酯前药的制备方法,包括如下步骤:
[0014]
(1)吉西他滨与tmscl反应保护羟基得到化合物a;化合物a在吡啶溶液中与dmtrcl反应保护氨基得到化合物b;化合物b在氟化铵作用下脱除硅烷保护基得到化合物c;
[0015]
(2)化合物c与三氯氧磷、乙醇酸甲酯在nmi的作用下反应得到化合物d;
[0016]
(3)化合物d在酸性溶液中脱除dmtr保护基,在成盐后得到所述吉西他滨环磷酸酯前药;
[0017]
其中,所述化合物a、化合物b、化合物c和化合物d的结构分别为:
[0018][0019]
进一步的:所述酸性溶液包括盐酸、硫酸、甲酸、乙酸、硝酸、甲磺酸、磷酸、对甲苯磺酸、三氟乙酸、焦谷氨酸、樟脑磺酸、马来酸、柠檬酸、乳酸、草酸、苹果酸、酒石酸、羟基马来酸、苯乙酸、谷氨酸、苯甲酸、水杨酸、对氨基苯磺酸、2-乙酰氧基-苯甲酸、富马酸、乙烷二磺酸、丙二酸、琥珀酸、抗坏血酸、1,2-乙烷二磺酸、2,4-二羟基苯甲酸、α-酮戊二酸、二氯乙酸、1-羟基-2-萘甲酸
[0020]
进一步的:所述步骤(3)中成盐后所成的盐包括盐酸盐、硫酸盐、甲酸盐、乙酸盐、硝酸盐、甲磺酸盐、磷酸盐、对甲苯磺酸盐、三氟乙酸盐、焦谷氨酸盐、樟脑磺酸盐、马来酸盐、柠檬酸盐、乳酸盐、草酸盐、苹果酸盐、酒石酸盐、羟基马来酸盐、苯乙酸盐、谷氨酸盐、苯甲酸盐、水杨酸盐、对氨基苯磺酸盐、2-乙酰氧基-苯甲酸盐、富马酸盐、乙烷二磺酸盐、丙二酸盐、琥珀酸盐、抗坏血酸盐、1,2-乙烷二磺酸盐、2,4-二羟基苯甲酸盐、α-酮戊二酸盐、二氯乙酸盐、1-羟基-2-萘甲酸盐。
[0021]
本发明还提供了所述的吉西他滨环磷酸酯前药在用于制备抗肿瘤药物中的应用。
[0022]
进一步的:所述肿瘤包括胰腺癌、前列腺癌、肝癌、胃癌、结肠癌、肺癌、乳腺癌、卵巢癌、黑色素瘤、胆管癌、脑胶质瘤、肾癌、鼻咽癌、尿路上皮癌、骨髓瘤、套细胞淋巴瘤、霍奇金淋巴瘤、白血病等所有人类癌症。
[0023]
进一步的:所述吉西他滨环磷酸酯前药为zl-20、zl-22、zl-34、zl-42、zl-64、zl-65、zl-66、zl-67、zl-68。
[0024]
本发明还提供了一种药物组合物,其将吉西他滨环磷酸酯前药,或其光学异构体、非对映异构体、消旋体或三者的混合物,或其药学上可接受的盐、溶剂合物、氘代物作为有效成分。
[0025]
与现有技术相比,本发明的优点和有益效果是:
[0026]
本发明通过简单的方法制备得到的吉西他滨环磷酸酯前药,不仅结构新颖,且经药理学证实,其克服了核苷类抗肿瘤药物容易出现耐药性的问题,对多种肿瘤细胞具有很好的抑制作用,因此具有显著的抗肿瘤作用,尤其是zl-22和zl-42在人血浆中具有很好的稳定性,能够提高吉西他滨环磷酸酯前药的应用性,所以吉西他滨环磷酸酯前药对肿瘤的治疗具有巨大的应用前景。
附图说明
[0027]
图1是zl-22的核磁共振氢谱;
[0028]
图2是zl-22的ms谱;
[0029]
图3是zl-34的核磁共振氢谱;
[0030]
图4是zl-34的ms谱;
[0031]
图5是zl-42的核磁共振氢谱;
[0032]
图6是zl-42的ms谱;
[0033]
图7是zl-64的核磁共振氢谱;
[0034]
图8是zl-64的ms谱;
[0035]
图9是zl-22在人血浆中的稳定性测试结果;
[0036]
图10是zl-42在人血浆中的稳定性测试结果。
具体实施方式
[0037]
结合以下具体实例对本发明的技术方案作进一步详细的说明。但本发明不限于下述实施例。下述实施例中,如无特殊说明,所使用的实验方法均为常规方法,所用材料、试剂等均可从生物或化学试剂公司购买。
[0038]
实施例1
[0039]
化合物c的合成路线如下:
[0040][0041]
实验步骤:
[0042]
盐酸吉西他滨(1g,3.34mmol,1eq)悬浮于18ml无水吡啶中。在-5℃下,tmscl
(2.17g,20mmol,6eq)慢慢地滴加到上述反应体系中,搅拌1h,升温到室温搅拌30min。tlc监测反应完全。直接加入dmap和dmtrcl,体系加热到55℃过夜反应。蒸干溶剂,用甲苯带干溶剂,加入二氯甲烷(dcm)和水,分层后,dcm层用1n的柠檬酸洗涤,饱和碳酸氢钠洗涤,盐水洗涤,无水硫酸钠干燥,过滤蒸干得2.7g,直接用于下一步反应。
[0043]
上述产物(2.4g,3.4mmol)溶解于30ml甲醇中,加入氟化铵(377.4mg,10.2mmol,3eq),加热到(55-60)℃反应30mim,tlc监测反应完全,蒸干有机溶剂,加dcm和水洗涤,柱层析纯化得1.2g淡黄色固体c。
[0044]
氢谱:1h nmr(500mhz,dmso-d6)δ8.52(s,1h),7.59(d,j=7.6hz,1h),7.27(t,j=7.5hz,2h),7.23
–
7.15(m,3h),7.13(d,j=8.9hz,4h),6.84(d,j=8.7hz,4h),6.29(d,j=7.6hz,1h),6.20(d,j=6.6hz,1h),5.98(t,j h-2f=8.2hz,1h,),5.14(t,j=5.2hz,1h),4.16-4.05(m,1h),3.72(d,j=12.7hz,9h),3.63
–
3.51(m,1h).lcms(esi+):[m+na]
+
found 588.07.99.45%purity.calcd for c
30h29
f2n3o6:565.20.
[0045]
实施例2
[0046]
化合物d的合成路线如下:
[0047][0048]
实验步骤:
[0049]
三氯氧磷(337mg,2.2mmol,3eq)溶于3ml的dcm中,并置于-15℃的冷井中。乙醇酸甲酯和三乙胺溶于无水dcm中后慢慢地滴加到上述反应中,反应搅拌3h后,产生白色沉淀,过滤备用。化合物c(360mg,0.64mmol,1eq)、三乙胺(3eq)、nmi(5eq)溶于无水的dcm中,将过滤的磷酸酯溶液的滤液慢慢地滴加入体系中,滴加完毕后室温搅拌1-2h。tlc检测反应完全后prep-tlc制备得200mg化合物d,收率44.7%。
[0050]
氢谱:1h nmr(400mhz,dmso-d6)δ8.67(s,1h),7.79
–
7.64(m,1h),7.31
–
7.24(m,2h),7.21-7.18(m,3h),7.12(d,j=8.8hz,4h),6.88(d,j=8.8hz,4h),6.45-6.25(m,2h),5.42
–
4.94(m,1h),4.87-4.62(m,4h),4.49
–
4.25(m,1h),3.74-3.71(m,9h).lcms(esi-)[m-h]-:found 698.19.calcd for c
33h32
f2n3o
10
p:699.18.
[0051]
实施例3
[0052]
化合物zl-22的合成路线如下:
[0053][0054]
实验步骤:
[0055]
化合物d(560mg,0.8mmol,1eq)溶于5-10ml的dcm中,加入三乙基硅烷(650mg,5.6mmol,7eq),室温下慢慢地滴加cf3cooh(912mg,8mmol,10eq),体系颜色变红,反应30min后红色慢慢地褪去。tlc检测原料全部反应完全,蒸干溶剂,用dcm带一遍,加入少量二氯和甲醇溶解产物,加入乙醚后析出絮状固体,抽滤得白色固体200mg,即为化合物zl-22。
[0056]
参见图1和图2:
[0057]
氢谱:1h nmr(400mhz,dmso-d6)δ8.78(s,1h),8.45(d,j=10.0hz,1h),8.07-7.87(m,1h),6.42(s,1h),6.06
–
5.93(m,1h),5.50
–
5.13(m,1h),4.86
–
4.78(m,3h),4.73-4.63(m,1h),4.55-4.49(m,1h),4.42-4.30(br,1h),3.83
–
3.60(m,3h).
[0058]
磷谱:
31
p nmr(162mhz)δ-4.88(s,1h),-6.85(s,1h).
[0059]
lcms(esi+):[m+h]
+
found 397.85.calcd for c
12h14
f2n3o8p:397.05.
[0060]
实施例4
[0061]
化合物zl-20的合成路线如下:
[0062][0063]
实验步骤:
[0064]
zl-22(200mg,0.5mmol)溶于5-10ml的甲醇溶液中,加入氢氧化钠(20mg,0.5mmol)的水溶液,室温搅拌2-4h。tlc检测原料全部反应完全,蒸干甲醇溶剂,制备纯化得白色固体120mg,即为化合物zl-20。
[0065]
lcms(esi+):[m+h]
+
found 383.90.calcd for c
11h12
f2n3o8p:383.03.
[0066]
最后,zl-34、zl-42、zl-64、zl-65、zl-66、zl-67、zl-68的合成路线与zl-22一致。
[0067]
zl-34的结构表征如下:
[0068][0069]
参见图3和图4:
[0070]
氢谱:1h nmr(400mhz,dmso-d6)δ8.58(s,1h),8.24(s,1h),8.00-7.90(m,1h),6.41(s,1h),5.99-5.96(m,1h),5.50-5.31(m,1h),4.85
–
4.73(m,2h),4.71-4.62(m,1h),4.54-4.48(m,1h),4.39
–
4.32(m,1h),4.22-4.16(m,2h),1.22(t,j=7.1hz,3h).
[0071]
磷谱:
31
p nmr(162mhz)δ-4.84(s,1h),-6.81(s,1h).
[0072]
lcms(esi+):[m+h]
+
found 411.88.calcd for c
13h16
f2n3o8p:411.06.
[0073]
zl-42的结构表征如下:
[0074][0075]
参见图5和图6:
[0076]
氢谱:1h nmr(400mhz,dmso-d6)δ8.45(s,1h),8.15(s,1h),7.99-7.86(m,1h),7.47-7.30(m,5h),6.41(s,1h),5.96-5.93(m,1h),5.51-5.33(m,1h),5.26-5.19(m,2h),4.92
–
4.84(m,2h),4.82
–
4.71(m,1h),4.70
–
4.58(m,1h),4.53-4.46(m,1h).
[0077]
磷谱:
31
p nmr(162mhz)δ-4.86(s,1h),-6.83(s,1h).
[0078]
lcms(esi+):[m+h]
+
found 473.93.calcd for c
18h18
f2n3o8p:473.08.
[0079]
zl-64的结构表征如下:
[0080][0081]
参见图7和图8:
[0082]
氢谱:1h nmr(500mhz,cd3od)δ7.66(d,j=7.6hz,1h),6.40(s,1h),5.97(dd,j=7.6,1h),5.26-4.99(m,2h),4.91-4.88(m,0.5h),4.80
–
4.68(m,3h),4.68
–
4.62(m,0.5h),4.62
–
4.54(m,0.5h),4.37-4.27(m,0.5h),1.33-1.28(m,6h).
[0083]
磷谱:
31
p nmr(162mhz)δ-4.49(s,1h),-6.33(s,1h).
[0084]
lcms(esi+):[m+h]
+
found 425.98.calcd for c
14h18
f2n3o8p:425.08.
[0085]
实施例5:吉西他滨环磷酸酯前药的抗肿瘤细胞活性测定实验
[0086]
1、实验目的:
[0087]
测试送检化合物:gemcitabine、nuc-1031(阳性对照药)、zl-22、zl-34、zl-42、zl-64对肿瘤细胞株的半数抑制浓度ic
50
。
[0088]
2、实验方法:
[0089]
(1)细胞计数与铺板:
[0090]
观察细胞长满以后,吸出培养,加2ml pbs洗涤去残留的培养基,吸出pbs后,加0.5-1ml胰蛋白酶消化细胞4min,必要时放到培养箱中消化,加2ml培养液中和胰酶,用抢反复吹打溶液至细胞分散,吹打完毕后显微镜观察,细胞处于悬浮并相互独立不黏连,析出细胞悬浮液,在1000转/min离心4分钟,小心吸出培养基,保留底部细胞,加入4ml培养基,用枪反复吹打,保持细胞均匀,析出1.5ml培养基用于传代,剩余的细胞用细胞计数板计数后铺板。
[0091]
(2)化合物浓度配置与加药:
[0092]
配置化合物的dmso溶液20mm的储备液,40μl储备液加120μl dmso稀释成5mm。第一个孔配置30μmol 200μl(加入198.8μl培养基到第一个孔,加入1.2μl5mm的药到第一个孔中)。后面的加入每孔120μl培养基。第一个孔析出60μl到第二个孔,依次往后稀释(3倍稀释),稀释到第10个孔,留最后两个空做对照。药物稀释后,用排枪吸取50μl,加入到细胞板上,培养72h。72h后加入染色剂15μl/孔,培养2-4h,用酶标仪检测。
[0093]
3、实验结果:
[0094]
结果如表1所示,与阳性对照药nuc-1031相比,zl-22、zl-34、zl-42和zl-64的ic
50
均小于0.1μm,说明本发明的吉西他滨环磷酸酯前药对多种肿瘤细胞均具有较强的抑制作用,都优于阳性对照药nuc-1031。
[0095]
表1:检测样品对肿瘤细胞株的半数抑制浓度ic
50
[0096]
[0097][0098]a标准:a≤0.1μm;0.1μm≤b≤1μm;1μm≤c≤10μm
[0099]
实施例6:核磁共振测试zl-22和zl-42在人血浆稳定性实验
[0100]
1、实验目的:
[0101]
测试送检化合物zl-22和zl-42在人血浆中的稳定性。
[0102]
2、实验方法:
[0103]
zl-22(3mg)溶于0.3ml的pbs中,加入0.3ml人血浆,放置于37℃的水浴锅中,于0.5h、1h、3h、6h、9h、12h、24h、27h、30h置于400mhz的核磁共振仪上测试
31
p谱。
[0104]
zl-42(3mg)溶于0.05ml的dmso中,加入0.2ml pbs后变浑浊,再加入0.05ml的dmso变澄清,加入0.3ml人血浆,放置于37℃的水浴锅中,于1h、2h、3h、6h、8h、10h、12h、24h置于400mhz的核磁共振仪上测试
31
p谱。
[0105]
3、实验结果:
[0106]
zl-22和zl-42的核磁测试
31
p谱结果如图9-10所示,可以看出,zl-22和zl-42在人血浆中具有很好的稳定性。
[0107]
以上实施例仅用以说明本发明的技术方案,而非对其进行限制;尽管参照前述实施例对本发明进行了详细的说明,对于本领域的普通技术人员来说,依然可以对前述实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或替换,并不使相应技术方案的本质脱离本发明所要求保护的技术方案的精神和范围。