一种基于喹啉单元结构的5/6/6并环四齿环金属铂(ii)配合物磷光材料及应用
技术领域
1.本发明涉及一种发光材料及其应用,尤其涉及一种基于喹啉单元结构的5/6/6 并环四齿环金属铂(ii)配合物磷光材料及应用。
背景技术:
2.有机发光二极管(oled)和液晶显示器(lcd)相比较来说,它的优点是很 轻薄,颜色绚丽,色彩饱和度高,无需背景光源,可视角度和响应速度十分优秀。 预计随着之后oled材料与器件的发展和优化以及成本的缩减,oled将成为未 来显示与照明的主流之一。
3.oled领域的核心始终是发光材料的设计和开发。在传统的荧光发光材料中, 只有最多25%的单线态激子会被利用,其余的75%三线态激子会因跃迁禁阻的存 在而失活。磷光材料中的金属原子具有重原子效应,能够利用全部的单线态和三 线态激子,获得接近100%的激子利用率,所以说磷光材料对oled发光器件的效 率有着决定性的作用。我们可以通过设计不同类型的有机配体能够调节器件的发 光颜色与纯度、器件效率等性能。oled领域发展壮大的关键问题依然是新型磷光 发光材料的设计和开发,低的驱动电压和高外量子效率及长寿命的oled器件是 我们追求的最终目标。拥有喹啉类结构的配体,由于和金属形成配合物后,令其 原来非刚性非平面的结构转化为刚性平面结构,导致分子变形困难,其从激发态 回到基态时发生非辐射跃迁的速率显著降低,主要发生辐射跃迁,从而使金属配 合物发光。
4.目前能够满足商业化应用的重金属磷光有机配合物分子基本均为环金属铱(iii) 配合物分子,且数量有限。地壳中金属铂元素的含量和世界范围内的年产均为金 属铱元素的约十倍,用于制备铱(iii)配合物磷光材料的ircl
3.
h2o(1100人民币/克) 价格也要远高于制备铂(ii)配合物磷光材料的ptcl2(210人民币/克);此外,制备 铱(iii)配合物磷光材料时涉及含铱(iii)二聚体、铱(iii)中间体配体交换、mer-铱(iii) 配合物的合成和mer-到fac-铱(iii)配合物异构体转换四步反应,使总收率大为降低, 大大降低了原料ircl
3.
h2o的利用率,提高了铱(iii)配合物磷光材料的制备成本。 相比之下,铂(ii)配合物磷光材料的制备只有最后一步配体的金属化设计铂盐的反 应,铂元素利用率高,可进一步降低铂(ii)配合物磷光材料的制备成本。综上所述, 铂(ii)配合物磷光材料的制备成本要远低于铱(iii)配合物磷光材料。
5.基于双齿配体的环金属铂(ii)配合物分子刚性较低,两个双齿配体易扭曲、振 动而导致非辐射衰减,使其磷光量子效率低下;而基于三齿配体的环金属铂(ii)配 合物分子需要第二个配阴离子(如炔负离子、cl-、卡宾等),亦会导致配合物的化 学稳定性和热稳定性降低;上述原因均不利于其作为磷光材料在oled器件中的 应用。相比之下,基于四齿配体的环金属铂(ii)配合物分子刚性强、辐射发光速率 大、量子效率会大大提高,同时具有较高的化学稳定性和热稳定性,是发展新型 oled磷光材料的理想分子。而开发稳定而高效的新型磷光发光材料,对oled 产业的发展依然具有举足轻重的意义。
技术实现要素:
6.本发明针对现有技术不足,提供了一种基于喹啉单元结构的5/6/6并环四齿环 金属铂(ii)配合物磷光材料及应用。
7.为实现上述发明目的:本发明提供了:
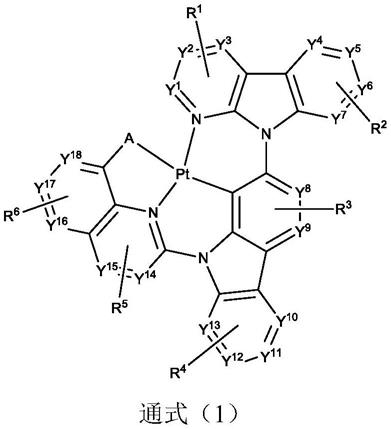
8.一种基于喹啉单元结构的5/6/6并环四齿环金属铂(ii)配合物磷光材料,其化学 式如通式(1)所示:
[0009][0010]
其中:
[0011]
a选自o,s或nr7;
[0012]
y1、y2、y3、y4、y5、y6、y7、y8、y9、y
10
、y
11
、y
12
、y
13
、y
14
、y
15
、 y
16
、y
17
和y
18
各自独立地为n或ch;
[0013]
所述r1、r2、r3、r4、r5、r6和r7的取代方式各自独立地表示为单取代、双 取代、三取代、四取代或者无取代;r1、r2、r3、r4、r5、r6和r7各自独立地表 示为氢、氘、烷基、卤代烷基、环烷基、烷氧基、芳基、杂芳基、芳氧基、卤素、 环烯基、杂环基、烯基、炔基、羟基、巯基、硝基、氰基、氨基、单或二烷基氨 基、单或二芳基氨基、酯基、腈基、异腈基、杂芳基、烷氧基羰基、酰氨基、烷 氧基羰基氨基、芳氧基羰基氨基、磺酰基氨基、氨磺酰基、氨基甲酰基、烷硫基、 亚磺酰基、脲基、磷酰胺基、亚胺基、磺基、羧基、肼基、甲硅烷基、取代的甲 硅烷基、聚合的基团、或其组合,且两个或者多个邻近的r1、r2、r3、r4、r5、 r6和r7可以选择性链接形成稠环。
[0014]
进一步地,所述通式(1)的结构式包括但不限于:
[0015]
[0016]
[0017]
[0018]
[0019]
[0020]
[0021]
[0022]
[0023]
[0024]
[0025]
[0026]
[0027]
[0028]
[0029]
[0030]
[0031]
[0032][0033]
进一步地,基于喹啉单元结构的5/6/6并环四齿环金属铂(ii)配合物磷光材料在 有机发光元件中的应用。所述有机发光元件为有机发光二极管、发光二极管或发 光电化学电池。
[0034]
进一步地,所述有机发光元件包括第一电极、第二电极及设置于所述第一电 极和所述第二电极之间的至少一个有机层;所述有机层包括基于喹啉单元结构的 5/6/6并环四齿环金属铂(ii)配合物磷光材料。
[0035]
本发明的有益效果是:与双齿配体铂配合物相比,本技术基于四齿配体的环 金属
铂(ii)配合物分子刚性强,可有效抑制由于分子振动而产生的非辐射跃迁,量 子效率会大大提高,同时四齿配体利于提高材料分子的化学稳定性和热稳定性。 本发明通过改变围绕金属中心的配体结构以及调控配体上的取代基结构来调节金 属铂配合物的光物理学性质。在oled显示和照明等诸多领域具有广阔的应用前 景。
附图说明
[0036]
图1为通过密度泛函理论(dft)计算得到的pt1与pt2、pt3、pt4、pt5、 pt6、pt7、pt8、pt27、pt28、pt30、pt32的最高分子占据轨道(homo)及最低 未占分子轨道(lumo)轨道分布比较图;
[0037]
图2为pt1在二氯甲烷溶液中室温下的光致发光光谱图;
[0038]
图3为有机发光元件的结构示意图;
[0039]
图4为pt1的高分辨质谱图。
具体实施方式
[0040]
以下对本发明的内容进行详细说明。以下所记载的构成要件的说明有时是基 于本发明的代表性实施方式或具体例而成,但本发明并不限定于此种实施方式或 具体例。
[0041]
本发明基于喹啉单元结构的5/6/6并环四齿环金属铂(ii)配合物磷光材料中所 包含的化合物具有下述通式(1)的结构。
[0042][0043]
通式(1)中配合物合成路线如下所示:
[0044][0045]
下面举例说明下述通式(1)代表的本发明的磷光材料的具体实例,然而,不解 释为限制本发明。
[0046]
除非另有说明,以下试验中所涉及到的所有商业试剂购买后直接使用,没有 进一步纯化。核磁共振氢谱均在氘代氯仿(cdcl3)或氘代二甲基亚砜(dmso-d6)溶 液中测得,氢谱使用400或500兆赫兹的核磁共振谱仪。如果用cdcl3作溶剂, 则氢谱以cdcl3(δ=7.26ppm)作为内标。如果用dmso-d6作溶剂,则氢谱以 dmso-d6(δ=2.50ppm)作为内标。以下缩写(或组合)用于解释氢谱峰:s=单峰, d=双重峰,t=三重峰,q=四重峰,p=五重峰,m=多重峰,br=宽峰。
[0047]
实施例1:四齿环金属铂(ii)配合物磷光材料pt1合成路线如下所示:
[0048][0049]
(1)中间体1-n的合成:向带有磁力转子的100ml三口瓶中,加入8-羟基 喹啉(5.00g,34.5mmol,1.00当量),n,n-二甲基甲酰胺40ml,然后将其置于低 温搅拌釜中,待温度降低后缓慢加入质量分数为60%的氢化钠(1.38g,34.5mmol, 1.00当量),置于常温后加入碘甲烷(5.14g,36.3mmol,1.05当量),反应过夜。 反应混合物加乙醇淬灭,减压蒸馏除去溶剂后,用乙酸乙酯萃取三次,合并有机 相,加无水硫酸钠干燥后,将所得粗品通过硅胶柱色谱进行分离纯化,淋洗剂(石 油醚/丙酮=2:1),得黑红色液体4.76g,静置固化,收率87%。1h nmr(400mhz, cdcl3):δ4.10(s,3h),7.06(d,j=8.0hz,1h),7.38
–
7.49(m,3h),8.13(d,j=8.0hz, 1h),8.94(dd,j=3.6hz,0.8hz,1h)。
[0050]
(2)中间体1-no的合成:向带有磁力转子的500ml三口瓶中加入1-n(5.40 g,34.2mmol,1.00当量),二氯甲烷170ml(1mmol/5ml),将三口瓶固定在低 温反应釜中降温后缓慢加入间氯过氧苯甲酸(8.84g,51.3mmol,1.50当量),取出 三口瓶,在室温下进行反应24小时。将反应混合物加入硫代硫酸钠淬灭,加水稀 释,后用二氯甲烷萃取4至5次,减压蒸馏除去溶剂,得粗品黑红色固体4.15g, 收率69%。直接用于下一步反应。
[0051]
(3)中间体1-br的合成:向带有磁力转子的1000ml三口瓶中依次加入中 间体1-no(6.90g,39.4mmol,1.00当量),四叔丁基溴化铵(19.05g,59.0mmol, 1.50当量)和分子筛,
再加入二氯甲烷700ml,在室温下搅拌10分钟后,加入 对甲苯磺酸酐(19.28g,59.0mmol,1.50当量),在室温下搅拌过夜,反应24小时。 将反应混合物过滤,用二氯甲烷洗涤滤渣,减压蒸馏除去溶剂,将所得粗品通过 硅胶柱色谱进行分离纯化,淋洗剂(石油醚/乙酸乙酯=10:1),得到白色固体4.3g, 收率46%。1h nmr(400mhz,cdcl3):δ4.06(s,3h),7.08(d,j=8.0hz,1h),7.37(d, j=8.0hz,1h),7.47
–
7.51(m,1h),7.55(d,j=8.8hz,1h),7.96(d,j=8.8hz,1h)。
[0052]
(4)中间体1-ome的合成:向带有磁力转子的封管中依次加入中间体1-br (571mg,2.4mmol,1.20当量),中间体aczczh(667mg,2.0mmol,1.00当量), 三(二亚苄基丙酮)二钯(55mg,0.06mmol,0.03当量),2-(二叔丁基膦)联苯(36mg, 0.12mmol,0.06当量)和叔丁醇钠(384mg,4.0mmol,2.00当量),抽换氮气三次, 在氮气保护下加入甲苯(8ml)。随后在110℃的油浴锅中搅拌反应2天,冷却至 室温,减压蒸馏除去溶剂,得到粗品。所得粗品用硅胶层析柱分离提纯,淋洗剂: 石油醚/乙酸乙酯的体积比为10:1-3:1,得到产物1-ome白色泡沫状固体293mg, 收率30%。1h nmr(400mhz,dmso-d6):δ3.81(s,3h),7.23(d,j=7.6hz,1h), 7.33
–
7.40(m,2h),7.45(t,j=7.2hz,1h),7.51
–
7.64(m,5h),7.66(dd,j=8.4,1.6hz, 1h),8.05(d,j=8.4hz,1h),8.09(d,j=8.0hz,1h),8.33(d,j=7.6hz,1h), 8.39
–
8.44(m,3h),8.53(d,j=8.0hz,1h),8.58(d,j=8.8hz,1h),8.67(dd,j=7.6, 1.6hz,1h)。
[0053]
(5)配体1-oh的合成:向带有磁力转子的50ml三口瓶中依次加入中间体 1-ome(264mg,0.54mmol,1.00当量)和吡啶盐酸盐(624mg,5.40mmol,10.00当 量),抽换氮气三次,在氮气保护下加入1,3-二甲基-2-咪唑啉酮(3ml)。随后在 180℃的油浴锅中搅拌反应1天,冷却至室温,加水用乙酸乙酯萃取三次,合并有 机相,之后再用饱和食盐水洗涤一次,并用无水硫酸钠干燥,减压蒸馏除去溶剂, 得到粗品。所得粗品用硅胶层析柱分离提纯,淋洗剂:石油醚/乙酸乙酯/二氯甲烷 的体积比为10:1:1-3:1:1,得到产物1-oh白色固体183mg,收率71%。1h nmr(500 mhz,dmso-d6):δ7.16(dd,j=7.5,1.5hz,1h),7.33
–
7.39(m,2h),7.43
–
7.59(m, 5h),7.67(dd,j=8.5,2.0hz,1h),7.71(d,j=8.5hz,1h),8.00(d,j=8.5hz,1h), 8.03(d,j=8.5hz,1h),8.31
–
8.32(m,2h),8.39(d,j=7.5hz,1h),8.42(dd,j=4.5, 1.5hz,1h),8.52(d,j=8.0hz,1h),8.57(d,j=9.0hz,1h),8.65(dd,j=7.5,1.5hz, 1h),9.72(s,1h)。
[0054]
(6)金属配合物pt1的合成:向带有磁力转子的50ml三口瓶中依次加入配 体1-oh(143mg,0.30mmol,1.00当量),四氯铂酸钾(131mg,0.315mmol,1.05当 量),四丁基溴化铵(10mg,0.03mmol,0.10当量),抽换氮气三次,在氮气保护 下加入醋酸(18ml),通氮气鼓泡30分钟。随后在120℃的油浴锅中搅拌反应2 天,冷却至室温,减压蒸馏除去溶剂,得到粗品。所得粗品用硅胶层析柱分离提 纯,淋洗剂为纯二氯甲烷,得到产物pt1橙黄色固体186mg,收率92%。产物在 氘代氯仿和氘代二甲基亚砜中的溶解度极差,无法得到完整的核磁数据。高分辨 质谱hrms(esi):c
32h18
n4opt[m+h]
+
计算值670.1201,实测值670.1202。
[0055]
光物理测试和理论计算说明:
[0056]
pt(ii)配合物使用titan软件包进行理论计算,利用密度泛函理论(dft)优化了 基态(so)分子的几何结构,使用b3lyp泛函进行dft计算,其中c、h、o和n原子 使用6-31g(d)基组,pt原子使用lanl2dz基组。
[0057]
实验数据及分析:
[0058]
由附图1可知,pt1配合物lumo与homo的能级差为3.07ev。pt1配合物 的lumo值为-1.74ev,其lumo大部分分布在氮杂咔唑上,只有很少一部分分 布在喹啉上。而pt1配合物的homo值为-4.81ev,其homo主要分布在喹啉、 氧原子和金属铂原子上。在喹啉上添加对叔丁基苯基和均三甲基苯基得到pt2和 pt3,对其进行dft计算发现,与pt1相比,它们的lumo和homo的分布没有 变化,而homo值有略微降低,homo的分布有一部分离散到了增加的对叔丁基 苯基和均三甲基苯基的苯环上。另外,将氧原子换成硫原子得到pt4,对其进行 dft计算发现,与pt1相比,它的lumo值有略微降低,homo值有略微升高, 从而能极差与pt1相比也减小了。通过观察pt4的lumo和homo的分布发现, 与pt1相比,它的homo的分布没有变化,而lumo则均匀分布在氮杂咔唑和 喹啉上。通过将配体上的碳原子换成氮原子得pt5、pt6、pt7、pt8,对其进行dft 计算发现,与pt1相比,lumo和homo的分布没有太大变化,lumo和homo 的值都有一定程度的降低。最后在配体的不同位置添加叔丁基得pt27、pt28、pt30、 pt32,对其进行dft计算发现,与pt1相比,lumo和homo的值与分布基本 没有变化。
[0059][0060]
由图2可知,铂配合物pt1在二氯甲烷溶液中可强烈发光,其最大发射波长 在619nm处,半峰宽为56nm,所以pt1可作为红光磷光材料;同时,其在约512nm 出也有以发射峰,故为具有双发射通道的发光材料;其溶液量子效率约50%。而 基于双齿配体的对比铂配合物ptq2的溶液量子效率则仅为1.5%左右(i.taydakov, k.lyssenko,r.saifutyarov,a.akkuzina,r.avetisov,a.mozhevitina,i.avetissov. dyes and pigments,2016,135,80-85.)。由此可知本技术基于四齿配体的铂配合物 相比双齿配体铂配合物的量子效率可大大提高。同时,由上述的理论计算可知, 可以通过对配体上的取代基进行调控,可使其衍生的金属配合物发光颜色蓝移或 进一步红移,使此类发光材料的光谱覆盖整个可见光区。
[0061]
本发明所述的含有喹啉结构单元的金属铂(ii)配合物发光材料在作为有机电致 发光器件的发光层中的应用。在有机发光元件中,从正负两电极向发光材料中注 入载子,产生激发态的发光材料并使其发光。通过通式(1)代表的本发明的化合物 可作为发光材料应用于有机光致发光元件或有机电致发光元件等有机发光元件。 有机光致发光元件具有在衬底上至少形成了发光层的结构。另外,有机电致发光 元件具有至少形成了阳极、阴极、及阳极和阴极之间的有机层的结构。有机层至 少包含发光层,可以仅由发光层构成,也可以除发光层以外具有1层以上的有机 层。作为这种其它有机层,可以列举空穴传输层、空穴注入层、电子阻挡层、空 穴阻挡层、电子注入层、电子传输层、激子阻挡层等。空穴传输层也可以是具有 空穴注入功能的空穴注入传输层,电子传输层也可以是具有电子注入功能的电子 注入传输层。具体的有机发光元件的结构示意如图3所示。在图3中,从下到上 共7层,依次表示衬底、阳极、空穴注入层、空穴传输层、发光层、电子传输层 和阴极,其中发光层为客体材料掺杂入主体材料的混合层。
[0062]
将实施例1中所表示的配合物作为磷光发光材料应用于oled器件,结构表 示为:
[0063]
ito/hatcn(10nm)/npd(65nm)/mcbp:实施例1中所表示的配合物(5-20 wt.%,20nm)/balq(10nm)/bpytp(30nm)/lif(1nm)/al(100nm)
[0064]
其中,ito为透明阳极;hatcn是空穴注入层,npd是空穴传输层,mcbp 是主体材料,实施例1中所表示的配合物(5-20wt.%是掺杂浓度,20nm是发光层 的厚度)是客体材料,balq是空穴阻隔层,bpytp为电子传输层,lif为电子注 入层,al为阴极。括号中单位为纳米(nm)的数字为薄膜的厚度。
[0065]
需要说明的是,所述结构为本发明发光材料的一个应用的举例,不构成本发 明所示发光材料的具体oled器件结构的限定,发光材料也不限于实施例1中所 表示的化合物。
[0066]
器件中应用材料的分子式如下:
[0067][0068]
本领域的普通技术人员可以理解,上述各实施方式是实现本发明的具体实施 例,而在实际应用中,可以在形式上和细节上对其作各种改变,而不偏离本发明 的精神和范围。例如,在不背离本发明的精神的情况下,这里描述的许多取代基 结构可以用其它结构代替。