1.本发明涉及化工技术领域,更具体地说,是涉及一种高断裂伸长率的聚酮及其合成方法。
背景技术:
2.聚酮(pok)是由一氧化碳与乙烯、丙烯等烯属不饱和烃交替共聚得到的共聚物,其中的co除了可以从煤造气获得,还可以从含co工业废气中通过净化获得,是一种具有优良综合性能的绿色高分子材料,可用作通用塑料、工程塑料、特种工程塑料、纤维和薄膜等材料。
3.聚酮材料具有耐磨、耐油、自润滑、耐冲击等诸多优良性能,耐冲击性比尼龙、pbt强2.3倍以上,耐磨性能是聚甲醛(pom)的10倍,对化学性质的稳定性比尼龙强1.4~1.5倍。聚酮材料的断裂伸长率是聚酮材料重要的性质之一,材料断裂伸长率的提高,意味着材料在收到外力作用断裂时吸收的能力越大,材料的力学性能就越好,抗冲击性能也在一定程度上增强。目前普通聚酮粉体产品断裂伸长率在150%~300%,悬臂梁抗冲击强度为6~15kj/m2。因此在不损失材料模量的前提下,提高产品韧性,增强聚酮材料的断裂伸长率等力学性能,将大大扩展材料的应用范围。
4.近年来,无机粒子加入聚合物中的报道日益增多,例如在公开号为cn103044769a的中国专利中通过加入超细碳酸钙作为填料,得到一种具有高强度、高断裂伸长率的聚丙烯材料;公开号为cn106633412a的中国专利通过添加纳米成核母粒得到高断裂伸长率、高韧性、高耐热性、高模量的聚丙烯组合物。无机粒子作为填料加入至聚合物材料中,能够提升材料的韧性、刚度、抗蠕变性等多种力学性能。其中,纳米级的无机粒子与传统无机粒子相比,其比表面积高,更接近聚合物基质的尺寸,可使聚合物的力学、光学性能等得到显著提高。但现有技术大多存在填料加入量较多、需复配增韧剂、混合不均匀、加工困难等缺陷。
技术实现要素:
5.有鉴于此,本发明的目的在于提供一种高断裂伸长率的聚酮及其合成方法,能够在不损失材料模量的同时,增加聚合物的韧性,从而提高聚酮粉体产品的断裂伸长率、抗冲击强度,赋予材料更优的力学性能。
6.本发明提供了一种高断裂伸长率的聚酮,所述高断裂伸长率的聚酮的断裂伸长率为500%~600%,悬臂梁缺口冲击强度为16kj/m2~25kj/m2。
7.本发明还提供了一种上述技术方案所述的高断裂伸长率的聚酮的合成方法,包括以下步骤:
8.a)一氧化碳和烯烃在加入催化剂、配体、无机纳米粒子及溶剂的密闭聚合反应釜中进行聚合反应,得到聚酮粉体产品;所述无机纳米粒子选自纳米sio2、纳米caco3和纳米al2o3的一种或多种。
9.优选的,步骤a)中所述无机纳米粒子的加入量为每100ml溶剂加入0.1g~1g。
10.优选的,步骤a)中所述催化剂包括硫酸钯、磺酸钯、硝酸钯和醋酸钯中的一种或多种。
11.优选的,步骤a)中所述配体为二齿配体,具有式(i)结构:
[0012][0013]
式(i)中,r1为至少含有3个碳原子的亚烷基,r2、r3、r4、r5独立地选自h、ch3、och3、oc2h4中的一种或多种。
[0014]
优选的,步骤a)中所述溶剂中还包括:
[0015]
醌和/或pka<6的酸的阴离子。
[0016]
优选的,所述醌包括苯醌和/或萘醌;
[0017]
所述pka<6的酸的阴离子包括硫酸根、对苯甲磺酸根、苯磺酸根、甲磺酸根、三氟甲磺酸根、高氯酸根、三氯乙酸根和三氟乙酸根中的一种或多种。
[0018]
优选的,步骤a)中所述一氧化碳和烯烃的摩尔比为1:(0.5~5)。
[0019]
优选的,步骤a)中所述聚合反应的温度为40℃~150℃,压力为2mpa~10mpa,时间为1h~12h。
[0020]
优选的,步骤a)中所述聚合反应为间歇式反应类型。
[0021]
本发明提供了一种高断裂伸长率的聚酮及其合成方法;所述高断裂伸长率的聚酮的断裂伸长率为500%~600%,悬臂梁缺口冲击强度为16kj/m2~25kj/m2;该合成方法包括以下步骤:a)一氧化碳和烯烃在加入催化剂、配体、无机纳米粒子及溶剂的密闭聚合反应釜中进行聚合反应,得到聚酮粉体产品;所述无机纳米粒子选自纳米sio2、纳米caco3和纳米al2o3的一种或多种。与现有技术相比,本发明提供的合成方法在反应之初将特定无机纳米粒子加入到聚合反应体系中,在不损失材料模量的同时,增加聚合物的韧性,从而有效提高聚酮粉体产品的断裂伸长率、抗冲击强度,赋予材料更优的力学性能。实验结果表明,相比于传统合成工艺,采用本发明提供的合成方法得到的聚酮粉体产品的断裂伸长率从300%提高至≥500%,悬臂梁缺口冲击强度由10kj/m2提升至20kj/m2左右。
附图说明
[0022]
图1为实施例1提供的聚酮产品的扫描电镜图;
[0023]
图2为对比例1提供的聚酮产品的扫描电镜图。
具体实施方式
[0024]
下面将结合本发明实施例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0025]
本发明提供了一种高断裂伸长率的聚酮,所述高断裂伸长率的聚酮的断裂伸长率为500%~600%,悬臂梁缺口冲击强度为16kj/m2~25kj/m2。本发明提供的高断裂伸长率的聚酮采用特定合成方法获得;该合成方法在反应之初将特定无机纳米粒子加入到聚合反应体系中,将其与聚酮粉体产品充分混合,在不损失材料模量的同时,增加了聚合物的韧性,从而提高了聚酮粉体产品的断裂伸长率,赋予材料更优的力学性能。
[0026]
需要指出的是,无机纳米粒子在本领域内的应用主要集中在聚合物后加工领域,将其作为填料加入至后加工体系中以提升某些性能,这在本文的摘要中也有提及;而本发明将无机纳米粒子在聚合反应之初加入,为本发明首创;同时与常规操作中将无机粒子作为后加工填料相比,本发明避免了现有技术存在的填料加入量较多、需复配增韧剂、混合不均匀、加工困难等缺陷,且纳米级粒子比表面积更高,更接近聚合物基质的尺寸,在聚合过程中与聚合物完全融合,提高了产品的断裂伸长率和缺口冲击强度;此外,本发明选用上述常见的无机纳米粒子,制备方法简单,生产成本低,易于后期进行工业化大规模生产。
[0027]
本发明还提供了一种上述技术方案所述的高断裂伸长率的聚酮的合成方法,包括以下步骤:
[0028]
a)一氧化碳和烯烃在加入催化剂、配体、无机纳米粒子及溶剂的密闭聚合反应釜中进行聚合反应,得到聚酮粉体产品;所述无机纳米粒子选自纳米sio2、纳米caco3和纳米al2o3的一种或多种。
[0029]
在本发明提供的合成方法中,首先将无机纳米粒子、催化剂和配体添加到溶剂中。在本发明中,所述无机纳米粒子为纳米sio2、纳米caco3和纳米al2o3的一种或多种,优选为纳米sio2、纳米caco3或纳米al2o3;所述无机纳米粒子的尺寸优选为10nm~100nm;本发明对所述无机纳米粒子的来源没有特殊限制,采用本领域技术人员熟知的市售商品即可。
[0030]
在本发明中,所述无机纳米粒子的加入量优选为每100ml溶剂加入0.1g~1g;具体可为0.1g:100ml、0.15g:100ml、0.2g:100ml、0.25g:100ml、0.3g:100ml、0.35g:100ml、0.4g:100ml、0.45g:100ml、0.5g:100ml、0.55g:100ml、0.6g:100ml、0.65g:100ml、0.7g:100ml、0.75:100ml、0.8g:100ml、0.85g:100ml、0.9g:100ml、0.95g:100ml或1g:100ml。
[0031]
在本发明中,所述催化剂优选包括硫酸钯、磺酸钯、硝酸钯和醋酸钯中的一种或多种,更优选为醋酸钯。本发明对所述催化剂的来源没有特殊限制,采用本领域技术人员熟知的上述钯催化剂的市售商品即可。
[0032]
在本发明中,所述催化剂在所述溶剂中的含量优选为0.01~1mmol/l,具体可为0.01mmol/l、0.15mmol/l、0.2mmol/l、0.25mmol/l、0.3mmol/l、0.35mmol/l、0.4mmol/l、0.45mmol/l、0.5mmol/l、0.55mmol/l、0.6mmol/l、0.65mmol/l、0.7mmol/l、0.75mmol/l、0.8mmol/l、0.85mmol/l、0.9mmol/l、0.95mmol/l或1mmol/l。
[0033]
在本发明中,所述配体优选为二齿配体,具有式(i)结构:
[0034]
[0035]
式(i)中,r1为至少含有3个碳原子的亚烷基,r2、r3、r4、r5独立地选自h、ch3、och3、oc2h4中的一种或多种。在此基础上,所述二齿配体具体可为2,2-二甲氧基-1,3-双[二(2-甲氧基苯基)膦基]丙烷、3,3-双-[双-(2-甲氧基苯基)膦甲基]-1,5-二氧杂-螺[5,5]十一烷和1,3-双-[二(2-甲氧基苯基)膦基]丙烷中的一种或多种。本发明对所述配体的来源没有特殊限制,采用本领域技术人员熟知的市售商品或自制品均可。
[0036]
在本发明中,所述配体与所述催化剂的摩尔比优选为(0.5~1.5):1,具体可为0.5:1、0.6:1、0.7:1、0.8:1、0.9:1、1:1、1.1:1、1.2:1、1.3:1、1.4:1或1.5:1。
[0037]
在本发明中,所述溶剂优选为低级脂肪族醇,包括但不限于甲醇、乙醇、异丙醇、乙二醇和丙三醇中的一种或多种。本发明对所述溶剂的来源没有特殊限制,采用本领域技术人员熟知的市售商品即可。
[0038]
在本发明中,所述溶剂中优选还包括:
[0039]
醌和/或pka<6的酸的阴离子;
[0040]
更优选还包括:
[0041]
醌和pka<6的酸的阴离子。
[0042]
在本发明中,所述醌优选包括苯醌和/或萘醌,更优选为苯醌或萘醌;其中,所述苯醌包括但不限于1,2-苯醌、1,4-苯醌和四氯对苯醌中的一种或多种。本发明对所述醌的来源没有特殊限制,采用本领域技术人员熟知的市售商品即可。
[0043]
在本发明中,所述醌与所述催化剂的摩尔比优选为(2~20):1,具体可为2:1、3:1、4:1、5:1、6:1、7:1、8:1、9:1、10:1、11:1、12:1、13:1、14:1、15:1、16:1、17:1、18:1、19:1或20:1。
[0044]
在本发明中,所述pka<6的酸的阴离子优选包括硫酸根、对苯甲磺酸根、苯磺酸根、甲磺酸根、三氟甲磺酸根、高氯酸根、三氯乙酸根和三氟乙酸根中的一种或多种,更优选为硫酸根、对苯甲磺酸根、苯磺酸根、甲磺酸根、三氟甲磺酸根、高氯酸根、三氯乙酸根或三氟乙酸根;所述阴离子优选由所述酸和/或所述酸对应的金属盐提供,所述酸和所述金属盐所提供的酸根离子可以相同也可以不同,所述酸包括但不限于硫酸、苯磺酸、对甲苯磺酸、甲磺酸、三氟甲基磺酸、高氯酸、三氯乙酸和三氟乙酸中的一种或多种,所述金属盐包括但不限于钠盐、钾盐、镁盐和铁盐中的一种或多种,具体可为三氟甲磺酸镁和/或三氟甲磺酸铁。
[0045]
在本发明中,所述酸与所述催化剂的摩尔比优选为(0.5~5):1,具体可为0.5:5、0.7:5、1:5、1.2:5、1.5:5、1.7:5、2:5、2.3:5、2.5:5、2.7:5、3:5、3.2:5、3.5:5、3.7:5、4:5、4.2:5、4.5:5、4.7:5或5:5;所述金属盐与所述催化剂的摩尔比优选为(0.5~20):1,具体可为0.5:1、1:1、1.5:1、2:1、2.5:1、3:1、3.5:1、4:1、4.5:1、5:1、6:1、7:1、8:1、9:1、10:1、11:1、12:1、13:1、14:1、15:1、16:1、17:1、18:1、19:1或20:1。
[0046]
在本发明提供的合成方法中,获得了含有上述各物质的溶剂后,向加入所述溶剂的反应釜内充入反应气体,进行聚合反应。在本发明中,所述溶剂的体积与所述反应釜的有效容积的比优选为(0.2~0.7):1,更优选为(0.3~0.5):1,具体可为0.3:1、0.4:1或0.5:1。
[0047]
在本发明中,所述反应气体包括一氧化碳和烯烃,所述烯烃包括但不限于乙烯和/或丙烯;所述一氧化碳和烯烃的摩尔比优选为1:(0.5~5),具体可为1:0.5、1:0.6、1:0.7、1:0.8、1:0.9、1:1、1:1.1、1:1.2、1:1.3、1:1.4、1:1.5、1:1.6、1:1.7、1:2、1:2.3、1:2.5、1:
2.7、1:3、1:3.2、1:3.5、1:3.7、1:4、1:4.2、1:4.5、1:4.7或1:5。
[0048]
在本发明中,所述聚合反应的压力优选为2mpa~10mpa,具体可为2mpa、2.5mpa、3mpa、3.5mpa、4mpa、4.5mpa、5mpa、5.5mpa、6mpa、6.5mpa、7mpa、7.5mpa、8mpa、8.5mpa、9mpa、9.5mpa或10mpa。所述聚合反应的压力由充入到反应釜内的反应气体提供,在本发明优选的实施例中,所述反应气体包括一氧化碳、乙烯和丙烯,其中,丙烯的充入量已提前计算好,通过控制一氧化碳和乙烯的充入量来调控聚合反应的压力。
[0049]
在本发明中,所述聚合反应温度优选为40℃~150℃,具体可为40℃、45℃、50℃、55℃、60℃、65℃、70℃、75℃、80℃、85℃、90℃、95℃、100℃、105℃、110℃、115℃、120℃、125℃、130℃、135℃、140℃、145℃或150℃;所述聚合反应的时间优选为1h~12h,具体可为1h、1.5h、2h、2.5h、3h、3.5h、4h、4.5h、5h、5.5h、6h、6.5h、7h、7.5h、8h、8.5h、9h、9.5h、10h、10.5h、11h、11.5h或12h。
[0050]
在本发明中,所述聚合反应优选为间歇式反应类型;所述聚合反应的过程优选在搅拌条件下进行;所述搅拌速度优选为100r/min~500r/min,具体可为100r/min、150r/min、200r/min、250r/min、300r/min、350r/min、400r/min、450r/min或500r/min。
[0051]
在本发明中,聚合反应结束后,得到的反应产物即为本发明合成的聚酮粉体;然后将所述聚酮粉体从反应溶剂中过滤、洗涤并干燥。其中,所述洗涤的洗剂优选为甲醇,所述干燥温度优选为70℃~90℃,具体可为70℃、75℃、80℃、85℃或90℃;所述干燥时间优选为1h~5h,具体可为1h、1.5h、2h、2.5h、3h、3.5h、4h、4.5h或5h。
[0052]
本发明采用上述合成方法,具有以下优点:
[0053]
(1)在反应之初将无机纳米粒子加入到聚合反应体系中,纳米级的无机粒子比表面积高,更接近聚合物基质的尺寸,将其与聚酮粉体产品充分混合,增加了聚合物的韧性,从而提高了聚酮粉体产品的断裂伸长率,赋予材料更优的力学性能。
[0054]
(2)将聚合物的后加工改性材料提前加入至聚合过程中,在不影响聚合反应的前提下,一方面克服无机纳米粒子在聚合物体系中的分散不良问题,使聚合物和无机纳米粒子混合更均匀,从而获得较理想的增韧效果;另一方面避免了填料使用量大,粉体加工生产不顺畅的问题,节省生产成本。
[0055]
实验结果表明,相比于传统合成工艺,采用本发明提供的合成方法得到的聚酮粉体产品的断裂伸长率从300%提高至≥500%,悬臂梁缺口冲击强度由10kj/m2提升至20kj/m2左右。
[0056]
本发明提供了一种高断裂伸长率的聚酮及其合成方法;所述高断裂伸长率的聚酮的断裂伸长率为500%~600%,悬臂梁缺口冲击强度为16kj/m2~25kj/m2;该合成方法包括以下步骤:a)一氧化碳和烯烃在加入催化剂、配体、无机纳米粒子及溶剂的密闭聚合反应釜中进行聚合反应,得到聚酮粉体产品;所述无机纳米粒子选自纳米sio2、纳米caco3和纳米al2o3的一种或多种。与现有技术相比,本发明提供的合成方法在反应之初将特定无机纳米粒子加入到聚合反应体系中,在不损失材料模量的同时,增加聚合物的韧性,从而有效提高聚酮粉体产品的断裂伸长率、抗冲击强度,赋予材料更优的力学性能。实验结果表明,相比于传统合成工艺,采用本发明提供的合成方法得到的聚酮粉体产品的断裂伸长率从300%提高至≥500%,悬臂梁缺口冲击强度由10kj/m2提升至20kj/m2左右。
[0057]
为了进一步说明本发明,下面通过以下实施例进行详细说明。本发明以下实施例
中所用的原料均为市售商品;聚酮粉体产品按照gb或者iso标准注塑成测试样条后测试性能:拉伸性能测试按照gb/t 1040.1和gb/t 1040.2测试,弯曲性能测试按照gb/t 9341测试(测试速度2mm/min),缺口冲击性能测试按照gb/t 1843测试;其他性能测试:粉体堆密度=聚合得到的聚酮质量/聚酮体积,熔体质量流动速度(mfr)测试按照gb/t 3682测试(测试温度和载荷:240℃/2.16kg),玻璃化转变温度、熔融温度测试按照gb/t 19466测试(dsc升温速率10k/min),负荷变形温度测试按照gb/t 1634.1测试(弯曲应力:0.45mpa,标准挠度:0.34mm,升温速率:120℃/h),硬度测试使用邵氏硬度。
[0058]
实施例1
[0059]
co/乙烯/丙烯三元聚合物的制备:
[0060]
在500ml高压反应釜中加入250ml甲醇,10.8mg对苯醌,16.5mg三氟甲磺酸镁,2ml三氟甲磺酸(7.5mmol/l),0.80g纳米sio2(粒径10nm~100nm),以及催化剂溶液:10ml甲醇,1.9mg醋酸钯,5.3mg 2,2-二甲氧基-1,3-双[二(2-甲氧基苯基)膦基]丙烷;加入上述物质后,高压釜内充入氮气保压、置换,然后充入丙烯15g,开始升温,设定温度95℃,持续充入co:c2h4=46:54的混合气,并维持反应压力为5mpa,搅拌速度400r/min,反应时间10h。
[0061]
反应结束后将所得聚酮粉体产品过滤,用甲醇洗涤,然后真空干燥箱80℃干燥3h,得到的产品量为95.03g;扫描电镜图参见图1所示。
[0062]
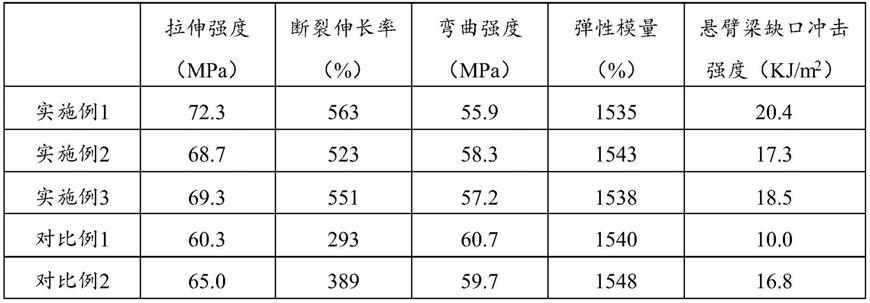
对本实施例制备的聚酮粉体产品进行样条注塑及性能测试,结果为:拉伸强度72.3mpa,断裂伸长率为563%,悬臂梁缺口冲击强度为20.4kj/m2。
[0063]
实施例2
[0064]
co/乙烯/丙烯三元聚合物的制备:
[0065]
在500ml高压反应釜中加入250ml甲醇,10.8mg对苯醌,16.5mg三氟甲磺酸镁,2ml三氟甲磺酸(6.5mmol/l),1.36g纳米caco3(粒径10nm~100nm),以及催化剂溶液:10ml甲醇,1.9mg醋酸钯,5.3mg 2,2-二甲氧基-1,3-双[二(2-甲氧基苯基)膦基]丙烷;加入上述物质后,高压釜内充入氮气保压、置换,然后充入丙烯15g,开始升温,设定温度95℃,持续充入co:c2h4=46:54的混合气,并维持反应压力为5mpa,搅拌速度400r/min,反应时间10h。
[0066]
反应结束后将所得聚酮粉体产品过滤,用甲醇洗涤,然后真空干燥箱80℃干燥3h,得到的产品量为92.63g。
[0067]
对本实施例制备的聚酮粉体产品进行样条注塑及性能测试,结果为:拉伸强度68.7mpa,断裂伸长率为523%,悬臂梁缺口冲击强度为17.3kj/m2。
[0068]
实施例3
[0069]
co/乙烯/丙烯三元聚合物的制备:
[0070]
在500ml高压反应釜中加入250ml甲醇,10.8mg对苯醌,16.5mg三氟甲磺酸镁,2ml三氟甲磺酸(6.5mmol/l),1.37g纳米al2o3(粒径10nm~100nm),以及催化剂溶液:10ml甲醇,1.9mg醋酸钯,5.3mg 2,2-二甲氧基-1,3-双[二(2-甲氧基苯基)膦基]丙烷;加入上述物质后,高压釜内充入氮气保压、置换,然后充入丙烯15g,开始升温,设定温度95℃,持续充入co:c2h4=46:54的混合气,并维持反应压力为5mpa,搅拌速度400r/min,反应时间10h。
[0071]
反应结束后将所得聚酮粉体产品过滤,用甲醇洗涤,然后真空干燥箱80℃干燥3h,得到的产品量为94.58g。
[0072]
对本实施例制备的聚酮粉体产品进行样条注塑及性能测试,结果为:拉伸强度
69.3mpa,断裂伸长率为551%,悬臂梁缺口冲击强度为18.5kj/m2。
[0073]
对比例1
[0074]
co/乙烯/丙烯三元聚合物的制备:
[0075]
在500ml高压反应釜中加入250ml甲醇,10.8mg对苯醌,16.5mg三氟甲磺酸镁,2ml三氟甲磺酸(6.5mmol/l),以及催化剂溶液:10ml甲醇,1.9mg醋酸钯,5.3mg 2,2-二甲氧基-1,3-双[二(2-甲氧基苯基)膦基]丙烷;加入上述物质后,高压釜内充入氮气保压、置换,然后充入丙烯15g,开始升温,设定温度95℃,持续充入co:c2h4=46:54的混合气,并维持反应压力为5mpa,搅拌速度400r/min,反应时间10h。
[0076]
反应结束后将所得聚酮粉体产品过滤,用甲醇洗涤,然后真空干燥箱80℃干燥3h,得到的产品量为87.56g;扫描电镜图参见图2所示。
[0077]
对本对比例制备的聚酮粉体产品进行样条注塑及性能测试,结果为:拉伸强度60.3mpa,断裂伸长率为293%,悬臂梁缺口冲击强度为10kj/m2。
[0078]
对比例2
[0079]
聚酮粉体与纳米sio2混合物的制备:
[0080]
按照对比例1中制备方法得到的聚酮粉体产品,取50g聚酮粉体、5g纳米sio2(粒径10nm~100nm)加入高速搅拌机中搅拌3~5min混合均匀,得到聚酮粉料与纳米sio2混合物。
[0081]
对本对比例制备的混合物进行样条注塑及性能测试,结果为:拉伸强度65.0mpa,断裂伸长率为389%,悬臂梁缺口冲击强度为16.8kj/m2。
[0082]
表1实施例与对比例的力学性能测试数据
[0083][0084]
表2实施例与对比例的其他性能测试数据
[0085][0086][0087]
通过对实施例和对比例进行比较可以看出,添加无机纳米粒子得到的聚酮粉体产品进行注塑样条测试中,在保证产品弹性模量及其他性能的同时,产品的断裂伸长率和悬
臂梁缺口冲击强度均有所提高,断裂伸长率从293%提升至500%以上,悬臂梁缺口冲击强度从10kj/m2提升至20kj/m2以上,产品的力学性能得到改善;扫描电镜图中,实施例1聚酮粉体产品颗粒均匀,对比例2中混合物存在混合不均匀、粉体结块的情况,注塑样条测试中产品的力学性能与不如实施例。
[0088]
所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。