1.本发明属于基因工程领域,具体地涉及一种土生隐球酵母编码脂质转运蛋白的基因rta1,以及转基因rta1酵母,以及它们在酸铝环境中的应用。
背景技术:
2.随着工业化的进展,大量的硫化物等物质排放在空气中,从而形成了酸雨,造成了许多地区存在土壤酸化的问题,如红壤地区。铝是地壳中含量最丰富的金属元素之一,约占地球总量的8%,其分布广泛而丰富。铝通常情况下以难溶性硅酸盐或氧化铝的形式存在,这些形态对生物毒性很微弱。随着酸雨现象的增加,土壤酸化的问题也越来越严重,土壤中铝离子的含量增加。其中在铝的各种化学形态中以无机单体铝(al
3+
)对各种农作物产生的毒害作用最显著,al
3+
的存在很大程度上取决于环境ph值(ph为3.0时al
3+
的含量为100%,ph为4.5时al
3+
的含量为80%,ph为6.0时al
3+
的含量则不到10%),所以铝毒是酸性土壤阻碍农作物生长的主要原因。铝能够影响生物的细胞膜结构,影响跨膜离子通道的活性,从而干扰细胞离子交换的稳态。有研究表明铝离子在土壤中的交换量约占阳离子的20-80%,导致了阳离子流失过多,从而造成土壤中各种营养元素的缺乏,如钾、钙、镁等,间接影响农作物对水分和营养成分的吸收,导致农作物产量的下降。
3.如何科学有效处理铝毒问题成为了世界上有待解决的难题,有研究发现用石灰来提高土壤的ph值,从而达到降低铝毒害的目的,但是这样容易导致土壤结构板结,土壤肥力下降,水分流失和土壤微生物减少等问题。在酸性的高铝土壤中,接种一些菌根真菌可以提高幼苗的存活率,其原因是菌根真菌活化了根际土壤中的难溶性磷酸盐,对游离态铝离子进行沉淀,从而减少铝毒对农作物的危害。菌根真菌提高植物耐铝作用的原因有:(1)菌根真菌通过菌丝生长而增加营养元素的吸收范围,体外排斥或分泌有机酸与al
3+
鳌合,使其沉淀,阻止其向植物根系运输等方式来减弱铝毒的影响;(2)菌根真菌的菌丝细胞壁的特殊结构可分离有毒金属离子,通过降低al
3+
含量达到减弱al
3+
对植物的伤害。所以有研究表明,在低ph和高铝环境中,植树造林的方面使用人工接种优良的外生菌根真菌的方法是最有效的对策。
4.在植物中,脂质转运蛋白在抗非生物胁迫中起着一定的作用。在小麦中脂质转运蛋白参与多种环境胁迫的响应,如低温、干旱、氧化胁迫等。在其他植物中,如玉米、拟南芥、芝麻等在面临上述的环境胁迫时,脂质转运蛋白的表达量也有所上升。真菌的脂质转运蛋白一般是由六个或者七个跨膜结构域组成的膜蛋白,跟动植物的脂质转运蛋白没有同源性,主要在真菌药物靶点方面有研究,而在真菌抗非生物胁迫方面未见报道。
技术实现要素:
5.本发明提供了一种脂质转运蛋白基因rta1,其核苷酸序列如seq id no:1所示,编码如seq id no:2所示氨基酸序列的蛋白质;其来源于土生隐球酵母(c. humicolus)bsll1-1菌株,基因序列全长1146bp,编码由381个氨基酸组成的脂质转运蛋白,分子量约为
42kda。
6.本发明另一目的是将上述脂质转运蛋白基因rta1应用在增强微生物抗酸胁迫或/和铝胁迫能力中。
7.为了实现本发明的上述目的,本发明提供了如下的技术方案:1、从云南省保山市龙陵县周边茶园茶树根际酸性土壤中分离的土生隐球酵母(c. humicolus)bsll1-1菌株中提取基因组dna,送上海人类基因组研究中心进行基因组测序,得到rta1基因序列。根据该序列设计引物,以土生隐球酵母cdna为模板pcr扩增rta1基因片段;将目的片段连接到pmd-19t载体上,得到含有目的片段的重组载体pmd19-t-rta1并热激转化到大肠杆菌感受态细胞dh5α中,涂布含氨苄青霉素的平板上,挑取阳性菌落测序,鉴定出编码rta1蛋白的基因,其序列如seq id no:1所示,该蛋白由381个氨基酸编码,氨基酸序列如seq id no:2所示;测序正确后通过酶切连接的方法构建pyes3-rta1重组载体;然后在invsc1感受态酵母细胞中加入pyes3-rta1重组载体,然后进行电击转化,将混合液涂于缺trp平板上筛选获得转基因rta1酵母。
8.2、rta1基因重组片段的构建将rta1基因的前1120bp核苷酸(rta1基因外的前1120位核苷酸)、后1080bp核苷酸(rta1基因外的后1080位核苷酸)和潮霉素基因三个独立片段分别设计扩增引物,前基因的反向引物与后基因的正向引物都带有一段同源臂;分别以rta1前臂和潮霉素基因为模板,扩增得到rta1前臂:前2/3潮霉素片段;以潮霉素基因与rta1后臂为模板,扩增得到后2/3潮霉素:rta1后臂重组片段;然后以上述两个重组片段为模板进行重叠延伸pcr,得到的重组片段rta1前臂:潮霉素基因:rta1后臂,命名为rta1f:hyg:rta1r;3、然后在土生隐球酵母感受态细胞中加入测序正确的重组片段rta1f:hyg:rta1r,然后进行电击转化,将混合液涂于含300μg/ml 潮霉素的ypd平板上筛选发生同源重组的土生隐球酵母突变体菌株;从潮霉素平板上挑取单菌落,用pcr和real-time pcr方法检测敲除是否成功;4、本发明比较了土生隐球酵母野生型菌株和土生隐球酵母突变型菌株、步骤1中的转基因rta1酵母和未转基因酵母在酸铝胁迫下的生长情况配制ph分别为3.5、4.5、5.5、6.5、7.5的gm固体培养基;配制ph4.5含铝离子的gm固体培养基,铝离子在gm固体培养基中的终浓度为50mmol/l、100mmol/l、150mmol/l;将土生隐球酵母野生型菌株、土生隐球酵母突变型菌株接种到ypd液体培养基中摇床培养过夜;按1%(v/v)接种量转接至新鲜的ypd液体培养基中,将菌液浓度调到od
600
为1;将菌液分别稀释至10-1
、10-2
、10-3
、10-4
,每个浓度菌液分别取1.5
µ
l接种到含有不同ph的gm固体平板上;在含铝离子的gm固体培养基上每个浓度菌液分别取2.5
µ
l接种,每种设置三个平板作为重复,在28℃下培养,观察不同菌株在酸铝胁迫下的生长情况;将初始od
600
=0.2的土生隐球酵母野生型菌株、土生隐球酵母突变型菌株接种于ph4.5、50mmol/l al
3+
的gm液体培养基中,28℃、200rpm培养,每隔2h测od
600
值,绘制生长曲线,以及在显微镜下观察ph4.5、不含铝离子的gm液体培养基中,ph4.5、铝离子终浓度为50mmol/l的gm液体培养基中土生隐球酵母菌株和突变菌株的菌体形态;
5、配制ph为4.5、7.5的gm诱导固体培养基;配制ph4.5含铝离子的gm诱导固体培养基,铝离子在gm固体培养基中的终浓度为0.1mmol/l、0.2 mmol/l、0.5 mmol/l;将invsc1酵母菌株、转空载invsc1菌株(i-p-t)、转rta1基因invsc1菌株(i-p-rta1)接种到ypd液体培养基中摇床培养过夜;按1%接种量转接至新鲜的ph4.5的gm诱导液体培养基中,将菌液浓度调到od
600
为1;将菌液分别稀释至100、10-1
、10-2
、10-3
,在不同ph的gm诱导固体平板上每个浓度菌液分别取1.5
µ
l接种;在含不同铝离子浓度的gm诱导固体平板上每个浓度菌液分别取2.5
µ
l接种,每种设置三个平板作为重复,在28℃培养箱中倒置培养,观察菌落大小,观察不同菌株在酸铝胁迫下的生长情况。
9.本发明的优点和技术效果如下:本发明为抵抗土壤中酸铝胁迫提供了一个基因rta1,rta1基因突变菌株对酸铝胁迫抗性明显下降,而在用一定浓度的铝离子以及不同ph环境处理转rta1基因invsc1菌株时,对转rta1基因invsc1菌株的生长情况影响不大。说明该基因具有抵抗酸铝胁迫的作用,该基因可以利用基因工程技术改造微生物用来抵抗环境中酸性和铝毒胁迫,本发明方法技术费用低,操作简便,对环境影响小,不会造成二次污染。
附图说明
10.图1为脂质转运蛋白基因rta1的扩增结果示意图,其中1泳道是rta1基因,m泳道是2000 marker;图2为rta1转基因酵母的pcr检测结果示意图,其中m泳道是2000 marker,2泳道为rta1转基因酵母;图3为不经铝处理的土生隐球酵母野生型菌株和土生隐球酵母野生型在50mmol/l铝离子浓度下rta1基因的表达结果;图4为本发明rta1f:hyg:rta1r重组基因片段的扩增示意图,其中左图中a为rta1前臂:前2/3潮霉素重组片段,b为后2/3潮霉素:rta1后臂重组片段;右图中c为rta1f:hyg:rta1r重组基因片段;图中m是dna marker; 图5为本发明rta1基因敲除的pcr和real-time pcr检测图,a图为敲除菌株的pcr检测图,其中1泳道是野生型菌株,2泳道是基因敲除菌株,m为dna分子量标准;b图为real-time pcr检测敲除菌株的基因转录情况,图中野生型-50是野生型菌株经50mmol/l al
3+
处理,突变体-50是转入重组片段的敲除菌株经50mmol/l al
3+
处理;图6为土生隐球酵母野生型菌株和rta1突变菌株抗酸胁迫能力检测结果;图7为土生隐球酵母野生型菌株和rta1突变菌株抗铝胁迫能力检测结果;图8为土生隐球酵母野生型菌株和rta1突变菌株经50mmol/l铝离子处理的生长曲线图;图9为本发明中未经处理的野生型和rta1突变体(上排),经50mmol/l铝离子处理的野生型和rta1突变体(下排)的镜检结果示意图;图10为转rta1基因invsc1菌株、invsc1酵母菌株、转空载invsc1菌株抗酸胁迫能力检测结果;图11为转rta1基因invsc1菌株、invsc1酵母菌株、转空载invsc1菌株抗铝胁迫能
力检测结果。
具体实施方式
11.下面通过实施例对本发明作进一步详细说明,但本发明的内容并不局限于此,本实施例中方法如无特殊说明的均按常规方法操作,所用试剂、菌种如无特殊说明的采用常规市售试剂或菌种,或按常规方法配置的试剂。
12.实施例1:土生隐球酵母(c. humicolus)bsll1-1菌株总rna提取及cdna的合成使用trizol试剂盒 (takara公司)进行酵母总rna的提取(冰上进行),步骤如下:取土生隐球酵母菌体0.1g,加入研钵中用液氮研磨至粉末状,然后加入1ml的trizol提取液继续研磨至清澈。将研磨液移到ep管,室温静置约5min,加入0.2ml氯仿颠倒混匀,无分化现象整体呈奶昔状,然后将样品置于冰上5min,12000rpm、4℃下离心10min;将上清转移至新的ep管中,再用0.2ml氯仿进行一次抽提。取上清并加入等体积异丙醇,-20℃ 静置1h后,12000 rpm、4℃下离心15min;弃上清,用75%的乙醇1ml清洗两次,12000rpm、4℃下离心5min,倒掉乙醇,自然晾干后用20-40
ꢀµ
l depc处理后的水进行溶解,-80℃保存。
13.用primescript
tm
rt reagent kit with gdna eraser反转录试剂盒对提取的总rna进行反转录;1、在管中配制下列模板rna/引物混合液;2、42℃保温2 min后迅速放在冰上;3、在上述管中配制下列转录反应液5、37℃保温15 min,85℃加热5s,然后放置在冰上,得到的cdna溶液可直接用于荧光定量pcr分析。
14.实施例2:rta1编码基因的克隆及测序以土生隐球酵母cdna为模板进行rta1基因的pcr扩增,扩增的引物为正向: aagcttatgcccgcaccccgcgcact(下划线为hindⅲ酶切位点),反向: ggatccttaccactggggaggggccttg(下划线为bamhⅰ酶切位点)。反应条件:94℃预变性3min,
然后94℃、30s,56℃、30s,72℃、1min进行35个循环,循环结束后72℃延伸5min;得到的pcr扩增产物进行琼脂糖凝胶电泳(图1),用dna胶回收试剂盒纯化目的条带;将目的片段连接到pmd19-t载体上,得到含有目的片段的重组载体pmd19-t-rta1;用热刺激法将其转化到大肠杆菌感受态细胞dh5α中,然后涂于含有氨苄青霉素的lb固体平板上,37℃倒置培养约12h;挑取单菌落于液体lb培养基中培养约12h后进行pcr验证,然后将扩增出目的片段的菌液用于提质粒,将重组载体送到昆明擎科生物有限公司测序,rta1基因核苷酸序列如序列表seq id no:1所示。
15.实施例3:rta1转基因invsc1酵母的构建将重组载体pmd19-t-rta1和pyes3/ct载体用hind
ꢀⅲ
和bamhⅰ进行双酶切,37℃酶切6h;然后将它们进行胶回收,配制连接体系:2
µ
l胶回收的rta1基因片段、3
µ
l胶回收的pyes3/ct载体、5
µ
l的solutionⅰ,将体系进行16℃过夜连接。用热刺激法将其转化到大肠杆菌感受态细胞dh5α中,然后涂于含有氨苄青霉素的lb固体平板上,37℃倒置培养约12h。挑取单菌落于液体lb培养基中培养约12h后进行pcr验证,然后将扩增出目的片段的菌液提质粒,在80ml invsc1感受态细胞中加入测序正确的20μl pyes3-rta1重组载体,置于冰上15min;然后将混合液加入到0.2cm冰预冷的电转杯中,进行电击(电击参数:25
ꢀµ
f、2500 v、200 ω),电击后加入400
µ
l预冷的山梨醇,在超净工作台将混合液均匀涂于缺trp的sd培养基平板上,置于28℃培养箱倒置培养2-3天;从缺trp平板上挑取单菌落,加入ypd液体培养基28℃培养过夜,收集菌体进行pcr检测;结果如图2所示,检测出1000到2000之间的条带。
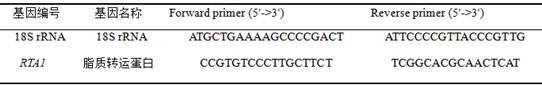
16.实施例4:荧光定量pcr分析土生隐球酵母rta1基因在50mmol/l铝胁迫下的表达设计土生隐球酵母的18s rrna和rta1基因的real-time pcr引物,18s rrna基因作为内参,引物序列如下:用sybr premix ex taqii(tli rnaseh plus)试剂盒进行real-time pcr,反应体系为20
µ
l(见下表),用applied biosystems 7500 fast real-time pcr system使用两步法pcr反应程序:stage 1:预变性,reps:1,95 ℃ 30 s;stage 2:pcr反应,reps:2,95 ℃ 5 s,60 ℃ 30s;反应结束后确认real-time pcr的扩增曲线和融解曲线,进行pcr定量时制作标准曲线等。
17.以18srrna基因为内参,以添加50mmol/l铝离子培养土生隐球酵母野生型的基因表达量为对照,应用2-δδct
法对实验结果进行分析,公式如下:(1)ct
处理
–
ct
内参(处理)
=δct
处理
;ct
对照
–
ct
内参(对照)
=δct
对照
;(2)δct
处理
–
δct
对照
=δδct;(3)倍数差异=2-δδct
;根据公式对照(ck)为“1”,并可算出不经铝处理的土生隐球酵母野生型和土生隐球酵母野生型在50mmol/l铝离子浓度下rta1基因的表达差异;铝胁迫下脂质转运蛋白基因rta1的real-timepcr分析结果表明,不经铝处理的土生隐球酵母的rta1基因不表达,经50mmol/l铝离子处理的土生隐球酵母高度表达脂质转运蛋白基因rta1(图3)。
18.实施例5:重叠pcr扩增rta1同源重组基因片段对rta1基因的1120bp、后1080bp核苷酸和潮霉素基因分别设计扩增引物f1和r1,f2和r2、f3和r3,其序列如下:f1::cactgcagaccatctccgac,r1:tgctccttcaatatcatcttctgtcggcggggtgtgtgtcgct;f2:ggatccacttaacgttactgaaatccacacattaactgtagtcc,r2:atctgcgcaacaaggtgg;f3:aaagttcgacagcgtctccg,r3:tccatacaagccaaccacgg。
19.下划线部分为同源臂。
20.扩增各个片段的重叠延伸pcr步骤如下:1、以脂质转运蛋白基因rta1前臂和潮霉素基因为模板,加入引物f1、r3在94℃,3min;94℃,30s;55℃,35s;72℃,2min进行35个循环,扩增rta1前臂:前2/3潮霉素重组片段;2、以潮霉素基因与rta1基因后臂为模版,加入引物f3、r2在94℃,3min;94℃,30s;55℃,35s;72℃,2min进行35个循环,扩增后2/3潮霉素:rta1后臂重组片段;3、以前两步得到的重组片段为模板进行重叠延伸pcr,先不加入引物在94℃,3min;94℃,30s;72℃,3min进行8个循环;然后加入引物f1、r2在94℃,3min、94℃,30s;55℃,35s;72℃,3min进行35个循环,扩增得到rta1f:hyg:rta1r重组基因片段(图4)。
21.实施例6:rta1基因的敲除菌株(突变型菌株)的构建在80μl土生隐球酵母感受态细胞中加入测序正确的20μlrta1f:hyg:rta1r重组片段,置于冰上15min;然后将混合液加入到0.2cm冰预冷的电转杯中,进行电击(电击参数:25
µ
f、2500v、200ω),电击后加入400
µ
l预冷的山梨醇,在超净工作台将混合液均匀涂于300μg
·
ml-1
潮霉素含量的ypd平板上,置于28℃培养箱倒置培养2-3d;从300μg
·
ml-1
潮霉素含量的ypd平板上挑取单菌落,加入ypd液体培养基28℃培养过夜,收集菌体进行pcr和real-timepcr检测;pcr结果显示(图5a),转入重组片段的菌株没有扩增出rta1基因,野生型扩增出rta1基因,这说明带有潮霉素基因的重组片段已经成功发生同源重组,转入重组片段的菌株中的rta1基因被成功敲除。real-timepcr检测结果表明(图5b),从50mmol/lal
3+
处理野生型和突变菌株的rta1基因的表达量来看,50mmol/lal
3+
处理突变菌株几乎没
有表达,这说明突变体中rta1基因已被敲除。
22.实施例7:土生隐球酵母突变菌株耐酸铝胁迫能力的检测在ph 4.5的gm固体培养基中加入铝离子让其终浓度为50mmol/l、100mmol/l、150mmol/l,以不加铝离子的固体培养基作为对照;同时配制ph分别为3.5、4.5、5.5、6.5、7.5的gm固体培养基;将野生型酵母和突变酵母接种到ypd液体培养基中摇床培养过夜,然后转接至新鲜的gm液体培养基中,将菌液浓度调到od
600
为1;然后稀释至10-1
、10-2
、10-3
、10-4
;每个浓度菌液分别取1.5
µ
l接种到含有不同ph的gm固体平板上;在含铝离子的gm固体培养基上每个浓度菌液分别取2.5
µ
l接种,每种设置三个平板作为重复,在28℃下培养,观察不同菌株在酸铝胁迫下的生长情况;结果见图6,图6结果显示在ph 3.5的gm固体培养基中突变体的生长受阻,而在ph 4.5-7.5的gm固体培养基中突变体跟野生型一样能够正常生长,但突变体菌落的长势比野生型弱;在50、100、150 mmol/l的铝离子处理下,突变体的生长情况不如野生型,而在150 mmol/l的铝离子处理下,突变体则无法生长(图7);将初始od
600
=0.2的土生隐球酵母野生型菌株、土生隐球酵母突变型菌株接种于ph4.5、50mmol/l al
3+
的gm液体培养基中,28℃、200rpm培养,每隔2h测od
600
值,绘制生长曲线(图8),在al
3+
终浓度为50mmol/l的gm培养基中,rta1突变体存在延滞期,rta1突变体延滞了24h;以及在显微镜下观察ph4.5、不含铝离子的gm液体培养基中,ph4.5、铝离子终浓度为50mmol/l的gm液体培养基中土生隐球酵母菌株和突变菌株的菌体形态(图9),从镜检结果可以看到在ph4.5、不含铝离子的gm液体培养基中rta1突变体与野生型相比较容易聚集成团;在ph4.5、铝离子终浓度为50mmol/l的gm液体培养基中rta1突变体与野生型的生长状况基本一致,没有菌体聚集现象。
23.实施例8:转rta1基因invsc1菌株耐酸铝胁迫能力的检测配制ph为4.5、7.5的gm诱导固体培养基;配制ph4.5含铝离子的gm诱导固体培养基,铝离子在gm固体培养基中的终浓度为0.1mmol/l、0.2 mmol/l、0.5 mmol/l;将invsc1酵母、转空载体酵母i-p-t、转rta1基因invsc1酵母i-p-rta1接种到ypd液体培养基中摇床培养过夜;按1%接种量转接至新鲜的gm诱导液体培养基中,将菌液浓度调到od
600
为1;然后稀释至100、10-1
、10-2
、10-3
;每个浓度梯度菌液取一定量的培养物接种到固体平板上,每种设置三个平板作为重复,在28℃培养箱中倒置培养,观察菌落大小;结果见10、11;insvc1、i-p-t在gm诱导固体培养基上生长受阻,且i-p-t在ph7.5的gm诱导固体培养基中无法生长,i-p-rta1在ph4.5和7.5的gm诱导固体培养基中则能正常生长(图10);在0.1、0.2、0.5mmol/l的铝离子处理下,与不加铝离子的相比随着铝离子浓度的升高,i-p-rta1的生长情况都比invsc1、i-p-t的生长情况要好。(图11)。