1.本发明属于医药技术领域,具体涉及一种硝唑尼特衍生物及其应用。
背景技术:
2.艰难梭菌(clostridium difficile)是梭菌属的一种专性厌氧菌,对氧十分敏感,很难分离培养,故得名,一般寄生在人的肠道内。通常艰难梭菌感染是由于过度服用某些抗生素,打破了肠内菌群的平衡,使得艰难梭菌的菌群生长速度加快,引发炎症。艰难梭菌会产生外毒素a和b,并在不同时期产生不同的作用。毒素a为肠毒素,在初期首先与黏膜细胞绑定,造成初级的破坏,可使肠壁出现炎症,细胞侵润,肠壁通透性增加,出血及坏死。毒素b为细胞毒素,损害细胞骨架,致细胞固缩坏死,直接损伤肠壁细胞导致腹泻。
3.艰难梭菌感染性疾病是由艰难梭菌菌体和/或艰难梭菌芽孢的感染引起的一种疾病。伪膜性肠炎是一种常见的艰难梭菌感染性疾病,临床表现为腹泻、腹痛、伴有全身中毒症状。症状突然开始,并伴随血压低,通常还伴有发烧,白细胞增多,甚至可导致死亡。此外,艰难梭菌菌体和/或艰难梭菌芽孢的感染还可能引起艰难梭菌感染性疾病并发症,常见的并发症包括肾盂肾炎、脑膜炎、腹腔及阴道感染、菌血症和气性坏疽等。近年来,艰难梭菌已成为引起医院内感染性疾病的重要病原菌,日益被人们所重视。
4.硝唑尼特:是一种硝噻柳酸酰胺的衍生物,其实际的作用机制虽尚不清晰,但被认为与抑制丙酮酸盐,铁氧化还原蛋白氧化还原酶的酶依赖性电子转移反应有关,后者对厌氧能量代谢至为重要。硝唑尼特孢子虫和肠贾第鞭毛虫之外,还对许多肠寄生虫,如贝氏等孢子虫、阿米巴原虫、人蛔虫、钩虫、毛首鞭虫、牛肉绦虫、短膜壳绦虫和肝片吸虫均有活性,目前也有报道公开硝唑尼特对甲硝唑治疗无效的艰难梭菌肠炎有好的疗效(musher dm,logan n,mehendiratta v,刘杨.甲硝唑治疗无效的艰难梭菌肠炎:硝唑尼特治疗有效[j].中国感染与化疗杂志,2008(01):46.)。
[0005]
大量的研究报道证实,硝唑尼特和多种硝唑尼特衍生物具有优异的生物活性,而其衍生物结构对生物活性有着不同的影响(张晗.硝唑尼特衍生物的合成及其抗菌定量构效关系研究[d].中国农业科学院,2012.),因此,进一步探索和合成结构相对简单,又具有更好的生物活性的硝唑尼特衍生物,为这类化合物的研究开辟新的领域,具有重要的意义。
技术实现要素:
[0006]
本发明的目的在于提供一种硝唑尼特的衍生物及其抑制艰难梭菌菌体的应用。
[0007]
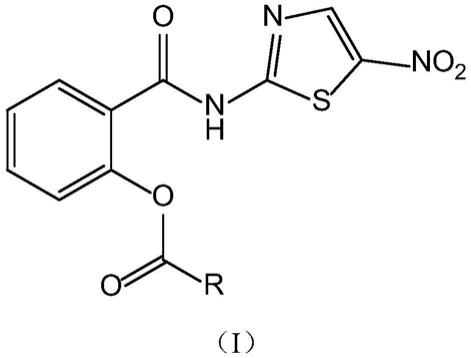
本发明提供了一种式(i)所示的化合物:
[0008][0009]
其中,r选自c
3-16
的直连或支链烷基。
[0010]
进一步地,上述化合物为:
[0011][0012]
本发明还提供了上述化合物在制备抑菌剂药物中的用途。
[0013]
进一步地,上述抑菌剂药物为抑制艰难梭菌菌体的药物。
[0014]
更进一步地,上述药物是预防和/或治疗艰难梭菌感染性疾病,和/或艰难梭菌感染性疾病并发症,和/或艰难梭菌感染性疾病的复发的药物。
[0015]
更进一步地,上述预防和/或治疗艰难梭菌感染性疾病并发症的药物是治疗和/或预防由艰难梭菌菌体感染引起的消化道感染综合征的药物。
[0016]
更进一步地,上述治疗和/或预防消化道感染综合征的药物为治疗和/或预防伪膜性肠炎、憩室炎、抗生素相关性腹泻、不全或完全性肠梗阻的药物。
[0017]
本发明提供了一种抑制艰难梭菌菌体的药物,所述药物是以上述化合物为活性成分,加上药学上可接受的辅料制备而成的制剂。
[0018]
进一步地,上述药学上可接受的辅料选自稀释剂、填充剂、着色剂、助流剂、润滑剂、粘合剂、稳定剂、助悬剂或缓冲剂的任一种或两种以上。
[0019]
更进一步地,上述制剂为口服制剂;优选地,所述口服制剂选自颗粒剂、胶囊剂、片剂、丸剂、混悬剂、乳剂。
[0020]
实验结果表明,本发明化合物能够有效抑制艰难梭菌的生长,相比于硝唑尼特和其它抗生素,在显著抑制艰难梭菌的活性的同时,对于肠道益生菌的影响也明显更小,且毒副作用更低,停药后不易复发。在制备预防和/或治疗艰难梭菌感染性疾病、抑制艰难梭菌
感染性疾病的复发,或治疗艰难梭菌感染性疾病并发症的药物中具有非常好的应用前景。
[0021]
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
[0022]
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
[0023]
图1为实验例3中各组金黄地鼠体重百分比曲线。
[0024]
图2为实验例3中各组金黄地鼠生存曲线。
具体实施方式
[0025]
本发明具体实施方式中使用的替唑尼特(tizoxanide)及其他化学试剂购自成都市科龙化工试剂厂。
[0026]
实施例1、本发明化合物i-1的合成
[0027][0028]
取2.0g替唑尼特(7.5mmol)于50毫升单口瓶中,加入20毫升乙酸乙酯和0.9g三乙胺(8.8mmol),搅拌下逐滴加入0.9g正丁酰氯(8.4mmol),滴加完后油浴加热至回流,反应2小时。tlc检测原料反应完全,降至室温,过滤除去固体,滤液用20毫升乙酸乙酯稀释,再分别用10毫升0.1m稀盐酸洗2次,10毫升10%碳酸氢钠溶液洗1次,最后用10毫升饱和食盐水洗至中性。乙酸乙酯层用2.5克无水硫酸钠干燥30分钟;过滤除去硫酸钠,滤液减压浓缩至干,所得粗品经硅胶柱层析精制,得化合物i-1。
[0029]1hnmr(400mhz,cdcl3)δ:。11.03(brs,1h),8.11-8.05(m,1h),8.02(d,j=7.9hz,1h),7.71(td,j=7.9,1.7hz,1h),7.46(t,j=7.6hz,1h),7.32(d,j=8.2hz,1h),2.69(t,j=7.4hz,2h),1.90-1.78(m,2h),1.06(t,j=7.4hz,3h)。
[0030]
esi-ms m/z:334.02[m-1]-。
[0031]
实施例2、本发明化合物i-2的合成
[0032][0033]
以替唑尼特和辛酰氯为原料,合成方法类似于实施例1,得到化合物i-2。
[0034]1hnmr(400mhz,cdcl3)δ:10.86(brs,1h),8.14(s,1h),8.04(dd,j=7.8,1.7hz,1h),7.75-7.66(m,1h),7.46(t,j=7.6hz,1h),7.31(d,j=8.2hz,1h),2.71(t,j=7.5hz,2h),1.85-1.73(m,2h),1.48-1.22(m,8h),0.90(t,j=6.8hz,3h)。
[0035]
esi-ms m/z:390.06[m-1]-。
[0036]
实施例3、本发明化合物i-3的合成
[0037][0038]
以替唑尼特和月桂酰氯为原料,合成方法类似实施例1,得到化合物i-3。
[0039]1hnmr(400mhz,dmso)δ:13.64(brs,1h),8.70(s,1h),7.82(d,j=7.7hz,1h),7.68(t,j=7.8hz,1h),7.44(t,j=7.5hz,1h),7.30(d,j=8.2hz,1h),2.55(t,j=7.1hz,2h),1.62-1.43(m,4h),1.32-1.16(m,14h),0.85(t,j=6.7hz,3h)。
[0040]
esi-ms m/z:446.10[m-1]-。
[0041]
实施例4、本发明化合物i-4的合成
[0042][0043]
以替唑尼特和十四烷酰氯为原料,合成方法类似实施例1,得到化合物i-4。
[0044]1hnmr(400mhz,cdcl3)δ:10.63(brs,1s),8.26-8.20(m,1h),8.08(d,j=7.8hz,1h),7.70(t,j=7.9hz,1h),7.47(t,j=7.5hz,1h),7.31(d,j=8.2hz,1h),2.72(t,j=7.5hz,2h),1.86-1.74(m,2h),1.47-1.38(m,2h),1.33-1.25(m,18h),0.90(t,j=6.7hz,3h)。
[0045]
esi-ms m/z:474.15[m-1]-。
[0046]
实施例5、制备本发明化合物的药用片剂组合物
[0047]
化合物i-1的药用片剂组合物,包含化合物i-1为1重量份,乳糖0.1-0.3重量份,淀粉0.4-0.2重量份,羧甲基淀粉钠0.008-0.014重量份,聚维酮k30适量,硬脂酸镁0.01-0.05重量份,40%乙醇0.5重量份;按照上述比例制备成片剂,即得本发明化合物i-1药用片剂,每片含化合物1-70为50-1500毫克。
[0048]
采用上述相同的方法,分别制得化合物i-2、i-3、i-4、i-5的药用片剂组合物。
[0049]
实施例6、制备本发明化合物药用胶囊剂组合物
[0050]
化合物i-2的药用胶囊剂组合物,含有300克化合物i-2、193克微晶纤维素、7克微粉硅胶,共计500克及2号空心胶囊;或者含有1200克化合物i-2、279克微晶纤维素、21克微粉硅胶、共计1500克及00号空心胶囊。制备方法为:
[0051]
a,将化合物i-2,微晶纤维素和微粉硅胶混合,得混合粉末;
[0052]
b,将混合粉末过120目筛后装填入胶囊并封口,共制1000粒。
[0053]
每粒胶囊含有化合物i-2的量为300毫克或者1200毫克。
[0054]
采用上述相同的方法,分别制得化合物1-1、i-3、i-4、i-5的药用胶囊剂组合物。
[0055]
实施例7、制备本发明化合物药用混悬剂组合物
[0056]
化合物i-3的药用混悬剂组合物,含有30克化合物i-3、50克阿拉伯胶、2克羧甲基纤维素钠、2克苯甲酸钠、3克枸橼酸、3克枸橼酸钠、20克蔗糖和890克水。制备方法为:
[0057]
a,将阿拉伯胶和羧甲基纤维素钠过100目筛后和水混合制成胶液并置于乳化器中;
[0058]
b,依次加入过100目筛的苯甲酸钠、枸橼酸、枸橼酸钠、化合物i-3和蔗糖并进行剪切制成。
[0059]
采用上述相同方法,分别制得化合物i-1、i-2、i-4、i-5的药用混悬剂组合物。
[0060]
以下通过实验例证明本发明化合物的有益效果。
[0061]
实验例1、本发明化合物对艰难梭菌体外活性的抑制
[0062]
(1)实验方法
[0063]
实验选取atcc bba1870、atcc 700057、atcc 630共3种艰难梭菌菌株,以添加补充剂的布式琼脂培养基测试。-80℃甘油冻存的菌株接到固体琼脂培养基上。放置37℃培养箱厌氧培养24~48h。本发明化合物的测试范围为32μg/ml-0.0156μg/ml,共11个两倍稀释浓度梯度;采用硝唑尼特作为对照化合物,万古霉素作为阳性对照药物。对照化合物硝唑尼特和阳性对照药物万古霉素测试范围为32μg/ml-0.0156μg/ml,所有对照化合物共11个两倍稀释浓度梯度。本发明化合物分别配置成一百倍测试浓度的高浓度工作液,当天使用,溶剂为100%dmso。对于每个稀释浓度琼脂板的准备,20μl高浓度工作液与2ml的熔化的、添加补充剂的布氏琼脂(45-55℃)混合并加入到六孔板里,待凝固。1%的dmso作为生长控制。实验当天,挑取适量单菌落悬浮于生理盐水,使用浊度仪将菌液浊度调节至od600=0.2,含约1
×
108cfu/ml。将此2μl菌液直接点在化合物固体稀释琼脂板中。因此六孔板每个孔含约105cfu艰难梭菌。此为测试板。将准备好的所有测试板置于35
±
2℃厌氧培养48h。培养48h后,通过肉眼观察,完全或显著抑制菌体生长的最低药物浓度为mic。
[0064]
(2)实验结果
[0065]
实验结果记录如下表1所示。结果表明,与对照化合物硝唑尼特相比,本发明的化合物对3种艰难梭菌菌体的抑制作用明显提高。本发明化合物均能够有效抑制艰难梭菌菌体的生长,对3种艰难梭菌菌体的mic≤1μg/ml,表现出了显著的抑菌活性,对atcc bba1870和atcc 630的mic值显著低于硝唑尼特和万古霉素,说明本发明化合物对艰难梭菌的抑菌活性显著优于硝唑尼特和万古霉素。
[0066]
表1本发明化合物抑制艰难梭菌菌体活性mic值(μg/ml)
[0067][0068]
实验例2、本发明化合物i-3对肠道益生菌的选择性
[0069]
(1)实验方法
[0070]
根据实验例1的结果,选取对各种艰难梭菌抑制效果最好的本发明化合物i-3进行对肠道益生菌选择性的验证。实验选取cgmcc no.1.5091两歧双歧杆菌、atcc19433粪肠球菌、atcc baa-835、atcc4356嗜酸乳杆菌、atcc19435乳酸乳球菌乳亚种、atcc7469鼠李糖乳杆菌、atcc11842保加利亚乳杆菌等共7种肠道益生菌菌株,以布式琼脂培养基测试。-80℃冻存的菌株接到固体琼脂培养基上。放置37℃培养箱厌氧培养24~48h。化合物i-3的测试范围为256μg/ml-0.25μg/ml,共10个两倍稀释浓度梯度;采用硝唑尼特、万古霉素、非达霉素作为对照化合物。三种对照化合物测试范围为128μg/ml-0.125μg/ml,所有对照化合物共10个两倍稀释浓度梯度。本发明化合物i-3配置成一百倍测试浓度的高浓度工作液,当天使用,溶剂为100%dmso。对于每个稀释浓度琼脂板的准备,20μl高浓度工作液与2ml的熔化的、添加补充剂的布氏琼脂(45-55℃)混合并加入到六孔板里,待凝固。1%的dmso作为生长控制。实验当天,挑取适量单菌落悬浮于生理盐水,使用浊度仪将菌液浊度调节至od600=0.2,含约1
×
108cfu/ml。将此2μl菌液直接点在化合物固体稀释琼脂板中。因此六孔板每个孔含约105cfu艰难梭菌。此为测试板。将准备好的所有测试板置于35
±
2℃厌氧培养48h。培养48h后,通过肉眼观察,完全或显著抑制菌体生长的最低药物浓度为mic。
[0071]
(2)实验结果
[0072]
实验结果记录如下表2所示。结果表明,与对照化合物硝唑尼特、万古霉素、非达霉素相比,本发明的化合物i-3在相同浓度下对7种肠道益生菌菌体的没有明显的抑制作用。与实验例1中抑制艰难梭菌菌体的mic相比,本发明化合物i-3的最大测试浓度远大于其对艰难梭菌菌体的mic值,说明本发明化合物i-3选择性的抑制了艰难梭菌菌体的生长,且选择性明显。即是说,硝唑尼特和万古霉素等抗生素在对艰难梭菌产生抑菌作用的同时还会对肠道益生菌的活性造成影响,而本发明化合物不但能够有效抑制艰难梭菌,而且还不影响肠道益生菌活性,具有显著的优势。
[0073]
表2本发明化合物抑制艰难梭菌菌体活性mic值(μg/ml)
[0074][0075][0076]
实验例3、本发明化合物对艰难梭菌体感染金黄地鼠的治疗
[0077]
(1)实验方法
[0078]
5周龄金黄地鼠60只,雄性,体重70-90g,北京维通利华实验动物技术有限公司购买。适应性喂养一周后随机分为6组,模型组和4个给药组口服给予10mg/kg克林霉素,空白组给予等体积生理盐水,记作第-1天。第0天,模型组和给药组给予艰难梭菌芽孢挑战,菌株为atcc 630,每只地鼠给予5e4个芽孢,空白组不作处理。第1天开始,4个给药组每天两次分别给予100mg/kg本发明化合物i-3、10mg/kg万古霉素、10mg/kg非达霉素以及100mg/kg硝唑尼特;空白组和模型组给予等量生理盐水,共给药20天,期间观察动物体重变化、腹泻和死亡情况。第21天起停药并继续观察至第45天。
[0079]
(2)实验结果
[0080]
各组金黄地鼠体重变化百分比记录如图1所示,各组金黄地鼠生存曲线记录如图2所示。模型组在感染艰难梭菌之后的5-10天内陆续死亡,最终全部死亡。各治疗组在前20天的治疗阶段中没有死亡,但在停药之后的5-10天之间,对照组硝唑尼特组和万古霉素组陆续出现地鼠死亡,最终硝唑尼特组存活4只,万古霉素组全部死亡。停药之后的15天非达霉素组也开始陆续死亡,最终全部死亡。化合物i-3组只在停药7天死亡一只,其他地鼠全部存活。与对照化合物万古霉素、非达霉素和硝唑尼特相比,本发明化合物i-3不仅在治疗阶段抑制艰难梭菌的生长,在停药之后也可以阻止艰难梭菌芽孢萌发带来的复发,说明本发明化合物对艰难梭菌感染金黄地鼠的治疗效果明显优于万古霉素、菲达霉素和硝唑尼特,具有非常好的临床应用前景。
[0081]
综上,本发明化合物能够有效抑制艰难梭菌的生长,相比于硝唑尼特和其它抗生素,在显著抑制艰难梭菌的活性的同时,对于肠道益生菌的影响也明显更小,且毒副作用更低,停药后不易复发。在制备预防和/或治疗艰难梭菌感染性疾病、抑制艰难梭菌感染性疾病的复发,或治疗艰难梭菌感染性疾病并发症的药物中具有非常好的应用前景。