1.本发明属于医药技术领域,具体涉及一种新型磷酸芦可替尼中间体其制备方法,以及磷酸芦可替尼的制备方法。
背景技术:
2.磷酸芦可替尼(ruxolitinib phosphate),由incyte公司和novartis合作研发,于2011年11月美国fda批准上市,成为首个获准的用于治疗骨髓纤维化的药物。
3.磷酸芦可替尼为抗肿瘤药。用于中危或高危的原发性骨髓纤维化(pmf)(亦称为慢性特发性骨髓纤维化),真性红细胞增多症继发的骨髓纤维化(ppv-mf)或原发性血小板增多症继发的骨髓纤维化(pet-mf)的成年患者,治疗相关疾病相关脾肿大或疾病相关症状。
4.其结构式如下所示:目前已报道的磷酸芦可替尼合成方法如下:方法1:文献ol.2009,11(9),1999-2002中以化合物2为起始物料,在手性小分子化合物4的催化下,与化合物3进行michael不对称加成得到化合物5,然后化合物5的醛基转化为氰基得到化合物6,最后化合物6脱除sem保护基并成磷酸盐得到目标产物1。关键手性中间体4是以手性小分子进行不对称诱导得到,该路线最大缺点在于手性诱导试剂分子量大,制备条件苛刻,制备成本高,不对称michael加成的选择性不高,不适合放大生产。
5.方法2:专利wo2007070514为芦可替尼化合物专利,该路线的最大缺点在于关键中间体7(sem保护的芦可替尼)需用手性制备柱制备,效率低,成本高,实际应用价值偏低。
6.方法3:文献angew.chem.int.ed.2015,54,7149
–
7153中以化合物2为起始原料,在金属铑与手性配体催化条件下与化合物3加成得到手性化合物中间体4,此路线起始原料化合物2不易得,中间体纯化困难、手性纯度不高,使用的贵金属催化剂成本高,不适合放大生产。
7.因此,开发一种反应条件温和、操作过程简单的方法,可顺利用于磷酸芦可替尼的工业化生产方法是目前本领域亟需解决的技术问题。
技术实现要素:
8.为解决上述问题,本发明公开了一种反应条件温和、操作过程简单的磷酸芦可替尼的制备方法。
9.为达到上述目的,本发明的技术方案如下:
(1)4-氯-7h-吡咯并[2,3-d]嘧啶被2-(三甲基甲硅烷基)乙氧基甲基(sem)保护,在溶剂中生成4-氯-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶;(2)4-氯-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶溶于溶剂中,与格氏试剂和硼酸酯反应生成化合物4-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶;(3)在溶剂中加入4-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶和4-溴-1h-吡唑,经过suzuki偶联生成4-(1h-吡唑-4-基)-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶;(4)在溶剂中加入4-(1h-吡唑-4-基)-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶、3-环戊基丙烯腈和碱,发生michael加成生成3-环戊基-3-(4-(7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)丙腈;(5)在溶剂中,将3-环戊基-3-(4-(7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)丙腈经过手性酸拆分后游离,得到(r)-3-环戊基-3-(4-(7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)丙腈;(6)在溶剂中,(r)-3-环戊基-3-(4-(7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)丙腈通过磷酸脱除保护基反应成盐,最终得到磷酸芦可替尼,即(r)-3-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-3-环戊基丙烯腈磷酸盐。
[0010]
其反应式如下所示:。
[0011]
进一步地,步骤(1)中,所述化合物1与2-(三甲基甲硅烷基)乙氧基甲基氯的物质的量之比为1.0:1.0~1.05,优选地,步骤(1)中,所述化合物1与2-(三甲基甲硅烷基)乙氧基甲基氯的物质的量之比为1.0:1.05;所述溶剂与化合物1的体积质量比为3.0~4.0:1.0,优选地,所述溶剂与化合物1的体积质量比为3.0:1.0;所述溶剂为n,n-二乙基苯乙酰胺;所述步骤(1)中的反应温度为20~25℃,反应时间为1~2h。
[0012]
进一步地,步骤(2)中,所述化合物2与格氏试剂与硼酸酯的的物质的量之比为1.0:1.0~1.05:1.0~1.05,优选地,步骤(2)中,所述化合物2与格氏试剂与硼酸酯的的物质的量之比为1.0:1.05:1.05;所述反应溶剂与化合物2的体积质量比为4.0~5.0:1.0,优选地,所述反应溶剂与化合物2的体积质量比为4.0:1.0。
[0013]
进一步地,步骤(2)中,所述反应的温度为0~5℃,反应的时间为1~2h;所述溶剂为二甲亚砜、n,n-二甲基甲酰胺的一种或两种,优选地,溶剂为二甲亚砜;所述格氏试剂为异丙基氯化镁、叔丁基氯化镁的一种或两种;优选地,使用的格氏试剂为异丙基氯化镁;使用的硼酸酯为联硼酸频那醇酯。
[0014]
进一步地,步骤(3)中,所述化合物3与化合物4的物质的量之比为1.0:1.0~1.1,优选地,步骤(3)中,所述化合物3与化合物4的物质的量之比为1.0:1.0;化合物3与有机金属催化剂的质量比为1:0.05~0.1,优选地,化合物3与有机金属催化剂的质量比为1:0.1;所述溶剂与化合物3的体积质量比为4.0~5.0:1.0,优选地,所述溶剂与化合物3的体积质量比为4.0:1.0。
[0015]
进一步地,步骤(3)中,所述suzuki偶联反应的温度为65~70℃,suzuki偶联反应的时间为1~2h;所述溶剂为二甲亚砜、n,n-二甲基甲酰胺的一种或两种;所述有机金属催化剂为ni(pcy3)2cl2;优选地,使用的溶剂为n,n-二甲基甲酰胺。
[0016]
进一步地,步骤(4)中,所述化合物5与化合物6的物质的量之比为1.0:1.0~1.2,优选地,步骤(4)中,所述化合物5与化合物6的物质的量之比为1.0:1.2;所述碱与步骤(4)中的化合物5的物质的量之比为0.2~0.5:1.0,优选地,碱与步骤(4)中的化合物5的物质的量之比为0.5:1;所述溶剂与化合物5的体积质量比为4.0~5.0:1.0,优选地,所述溶剂与化合物5的体积质量比为4.0:1.0;所述溶剂为n,n-二甲基乙酰胺;所述碱为2-甲基吡啶、4-二甲氨基吡啶中的一种或两种,优选地,碱为4-二甲氨基吡啶;所述michael加成反应的温度为25~30℃,michael加成反应的时间为6~7h。
[0017]
进一步地,步骤(5)中,所述化合物7与手性酸的物质的量之比为1.0:1.0~1.1,优选地,步骤(5)中,所述化合物7与手性酸的物质的量之比为1.0:1.0;所述溶剂与化合物7的体积质量比为 8.0~9.0:1.0,优选地,所述溶剂与化合物7的体积质量比为 8.0:1.0;进一步地,步骤(5)中,所述拆分的温度为55~60℃,拆分的时间为0.5~1h;所述溶剂为乙酸甲酯、乙酸异丙酯中的一种或两种,优选地,溶剂为乙酸甲酯;所述手性酸为d-(+)-樟脑酸、d-(+)-樟脑-10-磺酸中的一种或两种,优选地,手性酸 拆分剂为d-(+)-樟脑酸。
[0018]
进一步地,步骤(6)中,所述化合物8与磷酸的物质的量之比为 1.0:1.2~1.4,优选地,步骤(6)中,所述化合物8与磷酸的物质的量之比为 1.0:1.4;所述溶剂化合物8的体积质量比为20.0~22.0:1.0,优选地,所述溶剂与化合物8的体积质量比为20.0:1.0;所述溶剂为丙酮、1,4-二氧六环中的一种或两种,优选地,溶剂为丙酮;所述反应的温度为55~60℃,反应的时间为1~2h。
[0019]
与现有技术相比,本发明的有益效果为:本发明为制备磷酸芦可替尼提供了新的路线,该路线的各步反应均具有较高收率。
[0020]
该反应路线步骤(1) 化合物1与2-(三甲基甲硅烷基)乙氧基甲基氯的物质的量之
比优选为1.0:1.05,使用的溶剂与化合物1的体积质量比优为3.0:1.0,使用的溶剂选自n,n-二乙基苯乙酰胺,该条件下,该步收率可达95%。
[0021]
该反应路线步骤(2) 化合物2与格氏试剂与硼酸酯的的物质的量之比优选为1.0:1.05:1.05,反应溶剂与化合物2的体积质量比优选为4.0:1.0,使用的溶剂为优选二甲亚砜,使用的格氏试剂,优选异丙基氯化镁,使用的硼酸酯为联硼酸频那醇酯,该条件下,该步收率可达98%。
[0022]
该反应路线步骤(3) 所述化合物3与化合物4的物质的量之比优选为1.0:1.0,化合物3与有机金属催化剂的质量比优选为1:0.1,使用的溶剂优选为n,n-二甲基甲酰胺,溶剂与化合物3的体积质量比优选为4.0:1.0,使用的有机金属催化剂选自为ni(pcy3)2cl2,该催化剂稳定性相对较高,且相对常用的钯催化剂成本更低,该步收率可达95%。
[0023]
该反应路线步骤(4)化合物5与化合物6的物质的量之比优选为1.0:1.2,使用的碱,优选4-二甲氨基吡啶,碱与步骤(4)中的化合物5的物质的量之比优选为0.5:1,使用的溶剂选自n,n-二甲基乙酰胺,溶剂与化合物5的体积质量比优选为4.0:1.0,收率可达90%。
[0024]
该反应路线步骤(5)采用的酸性拆分试剂,优选d-(+)-樟脑酸,化合物7与手性酸的物质的量之比优选为1.0:1.0,溶剂优选乙酸甲酯,溶剂与化合物7的体积质量比优选为 8.0:1.0。此步骤拆分次数少,收率高,最高可达到45%。且拆分效果好,化合物8光学纯度可达99.8%,化学纯度可达99.5%。相对于其他专利使用的拆分试剂,本发明所述拆分试剂具有更高选择性,和更优的拆分效率。
[0025]
该反应路线步骤(6)使用的溶剂优选丙酮,化合物8与磷酸的物质的量之比优选为1.0:1.4,溶剂与化合物8的体积质量比为20.0:1.0。收率最高达95%,所得多批次磷酸芦可替尼成品光学纯度可达99.8%,化学纯度可达99.7%。
[0026]
该反应路线的后处理简单,无需柱层析,提高了制备的效率,适合工业化生产。
[0027]
且采用该路线,所需要的原料及用到的试剂都比较容易得到,与现有技术比,本发明方法更经济、更适于工业化生产。
[0028]
除特殊说明外,本发明所用试剂和原料均市售可得。本发明中化合物的中文命名与结构式有冲突的,以结构式为准;结构式有明显错误的除外。
附图说明
[0029]
图1为实施例1所得中间体化合物2的质谱图;图2为实施例3所得中间体化合物3的质谱图;图3为实施例4所得中间体化合物3的质谱图;图4为实施例5所得中间体化合物5的质谱图;图5为实施例7所得中间体化合物7的质谱图;图6为实施例9所得中间体化合物8的质谱图;图7为实施例11所得产物化合物9的质谱图;图8为实施例11所得产物化合物9的氢谱图;图9为实施例11所得产物化合物9的碳谱图。
具体实施方式
[0030]
下面结合附图和具体实施方式,进一步阐明本发明,应理解下述具体实施方式仅用于说明本发明而不用于限制本发明的范围。
[0031]
实施例14-氯-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶(化合物2)的制备先将若干4-氯-7h-吡咯并[2,3-d]嘧啶溶于3倍体积n,n-二乙基苯乙酰胺中,氮气保护,降温-10℃以下,分批加入氢化钠(1.05eq),加完后,升温至20~25℃,搅拌反应1~2h,然后控制温度-5℃以下,滴加干燥的2-(三甲基甲硅烷基)乙氧基甲基氯(1.05eq),滴完后,升温20~25℃,反应1~2h,tlc监测至结束反应,加入6倍体积饱和氯化铵溶液来淬灭反应。过滤析出的固体,滤饼用水及甲叔醚洗涤。40~45℃鼓风干燥,得4-氯-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶,收率95%,纯度≥94.0%。直接用于下一步反应。
[0032]
质谱图如图1所示,质谱数据如下:ms m/z:284.16[m+h]
+
。
[0033]
实施例24-氯-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶(化合物2)的制备先将若干4-氯-7h-吡咯并[2,3-d]嘧啶溶于4倍体积n,n-二乙基苯乙酰胺中,氮气保护,降温-10℃以下,分批加入氢化钠(1.05eq),加完后,升温至20~25℃,搅拌反应1~2h,然后控制温度-5℃以下,滴加干燥的2-(三甲基甲硅烷基)乙氧基甲基氯(1.0eq),滴完后,升温20~25℃,反应1~2h,tlc监测至结束反应,加入6倍体积饱和氯化铵溶液来淬灭反应。过滤析出的固体,滤饼用水及甲叔醚洗涤。40~45℃鼓风干燥,得4-氯-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶,收率91%,纯度≥91.0%。直接用于下一步反应。
[0034]
实施例34-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶(化合物3)的制备先将若干4-氯-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶溶于4倍体积二甲亚砜中,氮气保护,降温至-10℃以下,滴加异丙基氯化镁(1.05eq),加完后,升温至0~5℃下,搅拌反应1~2h,然后控制温度0~5℃以下,加入干燥的联硼酸频那醇酯(1.05eq),加完后,0~5℃反应1~2h,tlc监测至结束反应,加入6倍体积饱和氯化铵溶液来淬灭反应。料液中加入6倍体积乙酸乙酯萃取,水洗,饱和食盐水洗涤,无水na2so4干燥,过滤,减压浓缩得4-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶,收率98%,纯度≥95.0%。直接用于下一步反应。
[0035]
质谱图如图2所示,质谱数据如下:ms m/z:376.25[m+h]
+
。
[0036]
实施例44-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶(化合物3)的制备先将若干4-氯-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶溶于5倍体积n,n-二甲基甲酰胺中,氮气保护,降温至-10℃以下,滴加叔丁基氯化镁(1.0eq),加完后,升温至0~5℃下,搅拌反应1~2h,然后控制温度0~5℃以下,加入干燥的联硼酸频那
醇酯(1.0eq),加完后,0~5℃反应1~2h,tlc监测至结束反应,加入6倍体积饱和氯化铵溶液来淬灭反应。料液中加入6倍体积乙酸乙酯萃取,水洗,饱和食盐水洗涤,无水na2so4干燥,过滤,减压浓缩得4-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶,收率95%,纯度≥77.0%。直接用于下一步反应。
[0037]
质谱图如图3所示,质谱数据如下:ms m/z:376.27[m+h]
+
。
[0038]
实施例54-(1h-吡唑-4-基)-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶(化合物5)的制备先将若干4-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶溶于4倍体积n,n-二甲基甲酰胺中,加入醋酸钾(1.0eq)水溶液,4-溴-1h-吡唑(1.0eq),ni (pcy3)2cl2(10%),氮气保护,升温至65~70℃反应1~2h,tlc监测至结束反应。反应降至30℃以下,料液中加入6倍体积饱和食盐水,6倍体积乙酸乙酯,萃取,分液,无水na2so4干燥,过滤,减压浓缩得4-(1h-吡唑-4-基)-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶,收率95%,纯度≥99.0%。直接用于下一步反应。
[0039]
质谱图如图4所示,质谱数据如下:ms m/z:316.20[m+h]
+
。
[0040]
实施例64-(1h-吡唑-4-基)-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶(化合物5)的制备先将若干4-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶溶于5倍体积二甲亚砜中,加入醋酸钾(1.0eq)水溶液,4-溴-1h-吡唑(1.1eq),ni (pcy3)2cl2(5%),氮气保护,升温至65~70℃反应1~2h,tlc监测至结束反应。反应降至30℃以下,料液中加入6倍体积饱和食盐水,6倍体积乙酸乙酯,萃取,分液,无水na2so4干燥,过滤,减压浓缩得4-(1h-吡唑-4-基)-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶,收率92%,纯度≥94.0%。直接用于下一步反应。
[0041]
实施例73-环戊基-3-(4-(7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)丙腈(化合物7)的制备先将若干4-(1h-吡唑-4-基)-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶溶于4倍体积n,n-二甲基乙酰胺中,加入3-环戊基丙烯腈(1.2eq)、4-二甲氨基吡啶(0.5eq),25~30℃搅拌6~7h,tlc监测至结束反应。降温至0~10℃,加入8倍体积水,搅拌析晶0.5h,过滤,滤饼用水洗涤。40~45℃鼓风干燥得3-环戊基-3-(4-(7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)丙腈,收率90%,纯度≥99.0%。直接用于下一步反应。
[0042]
质谱图如图5所示,质谱数据如下:ms m/z:437.33[m+h]
+
。
[0043]
实施例83-环戊基-3-(4-(7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)丙腈(化合物7)的制备先将若干4-(1h-吡唑-4-基)-7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,
3-d]嘧啶溶于5倍体积n,n-二甲基乙酰胺中,加入3-环戊基丙烯腈(1.0eq)、2-甲基吡啶(0.2eq),25~30℃搅拌6~7h,tlc监测至结束反应。降温至0~10℃,加入8倍体积水,搅拌析晶0.5h,过滤,滤饼用水洗涤。40~45℃鼓风干燥得3-环戊基-3-(4-(7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)丙腈,收率87%,纯度≥96.0%。直接用于下一步反应。
[0044]
实施例9(r)-3-环戊基-3-(4-(7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)丙腈(化合物8)的制备将若干3-环戊基-3-(4-(7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)丙腈加入6倍体积乙酸甲酯中,搅拌溶解,25~30℃,向其中滴加d-(+)-樟脑酸(1.0eq)的2倍体积乙酸甲酯溶液。加完升温至55~60℃搅拌0.5~1h,降温至20~25℃搅拌2h。过滤,滤饼适量乙酸甲酯洗涤。
[0045]
洗涤后滤饼加入6倍体积乙酸甲酯中,加入三乙胺(0.5eq),25~30℃搅拌1~2h,过滤,滤饼适量乙酸甲酯洗涤,减压浓缩滤液得(r)-3-环戊基-3-(4-(7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)丙腈,收率45%,光学纯度≥99.8%,化学纯度≥99.5%。
[0046]
质谱图如图6所示,质谱数据如下:ms m/z:437.33[m+h]
+
。
[0047]
实施例10(r)-3-环戊基-3-(4-(7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)丙腈(化合物8)的制备将若干3-环戊基-3-(4-(7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)丙腈加入9倍体积乙酸异丙酯中,搅拌溶解,25~30℃,向其中滴加d-(+)-樟脑-10-磺酸(1.1eq)的2倍体积乙酸异丙酯溶液。加完升温至55~60℃搅拌0.5~1h,降温至20~25℃搅拌2h。过滤,滤饼适量乙酸异丙酯洗涤。
[0048]
洗涤后滤饼加入7倍体积乙酸异丙酯中,加入三乙胺(0.5eq),25~30℃搅拌1~2h,过滤,滤饼适量乙酸异丙酯洗涤,减压浓缩滤液得(r)-3-环戊基-3-(4-(7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)丙腈,收率42%,光学纯度≥99.5%,化学纯度≥99.4%。
[0049]
实施例11(r)-3-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-3-环戊基丙烯腈磷酸盐(化合物9)的制备将若干(r)-3-环戊基-3-(4-(7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)丙腈加入18倍体积丙酮中,搅拌溶解。20~30℃,向其中滴加磷酸(1.4eq)的2倍体积丙酮溶液。加完升温至55~60℃搅拌1~2h,降温至20~30℃搅拌2h,过滤,滤饼用适量丙酮洗涤。40~45℃鼓风干燥得(r)-3-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-3-环戊基丙烯腈磷酸盐,收率95%,光学纯度≥99.8%,化学纯度≥99.7%。
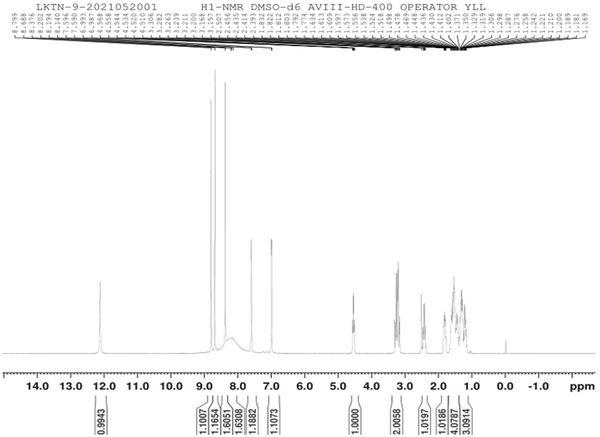
[0050]
氢谱图如图8所示,氢谱数据如下:1h-nmr(dmso-d6,400mhz) δ:12.150(1h,s),8.799(1h,s),8.688(1h,s),8.194-8.202(2h,m),8.140(2h,br),7.590-7.596(1h,m),
6.987-6.993(1h,m),4.510-4.568(1h,m),3.157-3.306(2h,m),2.393-2.507(1h,m),1.774-1.832(1h,m),1.371-1.634(4h,m),1.169-1.350(3h,m)。
[0051]
碳谱图如图9所示,碳谱数据如下:
13
c-nmr(dmso-d6,400mhz) δ:152.58,151.37,150.34,139.71,131.48,127.21,120.97,118.64,113.28,110.25,62.97,44.79,29.56,29.53,25.41,24.79,22.98。
[0052]
质谱图如图7所示,质谱数据如下:ms m/z:307.2[m+h]
+
。
[0053]
实施例12(r)-3-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-3-环戊基丙烯腈磷酸盐(化合物9)的制备将若干(r)-3-环戊基-3-(4-(7-((2-(三甲基硅基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)丙腈加入20倍体积1,4-二氧六环中,搅拌溶解。20~30℃,向其中滴加磷酸(1.2eq)的2倍体积1,4-二氧六环溶液。加完升温至55~60℃搅拌1~2h,降温至20~30℃搅拌2h,过滤,滤饼用适量1,4-二氧六环洗涤。40~45℃鼓风干燥得(r)-3-(4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)-3-环戊基丙烯腈磷酸盐,收率90%,光学纯度≥99.8%,化学纯度≥99.6%。
[0054]
需要说明的是,以上内容仅仅说明了本发明的技术思想,不能以此限定本发明的保护范围,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰均落入本发明权利要求书的保护范围之内。