1.本技术主张基于2020年2月27日申请的韩国专利申请10
‑
2020
‑
0023993号的优先权权益,且相应韩国专利申请的文献所揭示的所有内容均作为本说明书的一部分而包含在内。
2.本发明是有关于一种具有表面凸块的球状无机颗粒以及其制备方法,更具体而言是有关于一种形成有表面凸块且表面电荷得到控制的球状无机颗粒以及其制备方法。
背景技术:
3.无机颗粒在各种领域中用作原材料或最终制品,特别是用于化学催化、生物、半导体工程、钢化玻璃加工等广泛的范围。
4.合成这种无机颗粒的工程非常多样,合成方法根据制备接近方式分为进行组装原子的方法(自下而上(bottom up))与进行减小大块的大小的方法(自上而下(top down)),根据合成原理分为物理方法、机械方法、化学方法。在化学方法中,使用液相中的化学反应的液相反应法是作为陶瓷原料粉末合成法最广泛使用的方法。作为使用液相化学反应的粉末制备工程的种类,已知有溶胶
‑
凝胶法(sol
‑
gel method)、热分解法(pyrolysis method)、聚合络合法(polymerized complex method)、沉淀法(precipitation method)、水热合成法(hydrothermal method)等。
5.在合成无机颗粒的过程中,根据原子固有的组装特性来生长颗粒,并由此确定无机颗粒的最终形状。即,由于无机颗粒的形状是无机颗粒的固有性质,因此很难将相同成分的无机颗粒制备为不同的形状。
6.例如,二氧化铈(ceo2)晶体具有呈现出角的六边形结构的萤石(fluorite)颗粒形状。二氧化铈颗粒作为研磨颗粒被包含在半导体制备工程中用于化学机械研磨(chemical mechanical polishing,cmp)工程的浆料(slurry)中,且由于二氧化铈颗粒的呈现出角的结构而产生划痕(scratch)不良缺陷。因此,为了解决此问题,正在研究可将二氧化铈颗粒制备为球状的方法。然而,很难合成在将呈现出角的萤石(fluorite)结构的二氧化铈的形状改变为球状的同时,尺寸均匀且良好地分散的二氧化铈颗粒。
7.另外,根据无机颗粒的形状变化,颗粒的比表面积会产生差异,因此,颗粒表面上的化学反应程度可能会不同。例如,在使用无机颗粒作为催化的情况下,颗粒的比表面积与催化活性部位直接相关,且相对于相同体积比表面积大的颗粒的反应性优异。
8.无机颗粒的又一争议中的一者是分散稳定性。纳米尺寸的无机颗粒(以下,还称为“纳米颗粒”)通常在水溶液中在热力学上不稳定,且由于高的比表面积而难以稳定地分散。因此,存在的问题是在保管过程中可能出现颗粒的凝聚,因此形状或性质可能会发生变化。因此,需要一种用于提高纳米颗粒的分散性的方法。
9.由此,为了提高纳米颗粒的分散性,需要用于控制纳米颗粒的表面电荷的技术。特别是,例如在半导体cmp工程中用作浆料内研磨颗粒的二氧化铈或二氧化硅(silica)纳米颗粒在水溶液中的分散是非常重要的。因此,致力于通过调节浆料水溶液的ph以提供可在
研磨颗粒与膜质间产生更强引力的环境来提高研磨工程的效率。
技术实现要素:
10.发明要解决的问题
11.本发明要解决的问题是提供一种无机颗粒,所述无机颗粒具有球形形状而不是呈现出角的形状且水分散性优异,特别是对硅膜的研磨能力优异,同时划痕损伤少。
12.另外,本发明要解决的另一问题是提供一种制备所述无机颗粒的方法。
13.另外,本发明要解决的又一问题是提供一种将所述无机颗粒分散在水中的分散液。
14.用于解决问题的手段
15.本发明为了达成所述的技术问题,提供一种无机颗粒,所述无机颗粒为多个小颗粒凝聚形成的无机颗粒,所述小颗粒混合有结晶相与非晶相且结晶度为90%以下。
16.根据一实例,所述小颗粒的粒径可为10nm以下。
17.根据一实例,所述无机颗粒的密度为3.0g/ml至5.0g/ml,平均粒径为30nm至1000nm,粒径的标准偏差可为20以下。根据一实例,所述无机颗粒的等电点可为ph5至ph7。
18.根据一实例,所述无机颗粒在ph4的水分散液状态下可具有+30mv至+50mv或
‑
30mv至
‑
50mv的ζ电位。
19.根据一实例,所述无机颗粒可由选自由ga、sn、as、sb、ce、si、al、co、fe、li、mn、ba、ti、sr、v、zn、la、hf、ni及zr组成的群组中的一种以上的元素的氧化物形成。
20.根据一实例,所述无机颗粒为ceo2颗粒,ce
3+
/ce
4+
离子比可为40至60。
21.另外,本发明为了解决所述的另一技术问题,提供一种方法,所述方法包括以下步骤:(a)使自组装界面活性剂溶解于溶剂;(b)在实施所述(a)步骤之前、之后或同时,使无机物前驱体溶解或分散于所述溶剂来制备无机物前驱体溶液;以及(c)通过所述无机物前驱体与所述界面活性剂的自组装反应,在界面活性剂形成的壳中形成结晶相与非晶相混合的小颗粒,进而多个小颗粒凝聚形成无机颗粒。
22.根据一实例,所述自组装界面活性剂为选自具有能够与所述无机物前驱体结合的电荷的阳离子性界面活性剂、阴离子性界面活性剂及两性界面活性剂中的一种以上,且保有可进行缩合反应以及交联反应的官能基。
23.根据一实例,可进行缩合反应以及交联反应的所述官能基可为选自由酰胺基、硝基、醛基及羰基组成的群组中的一种以上。
24.根据一实例,所述自组装界面活性剂可为下述化学式1的高分子。
25.[化学式1]
[0026][0027]
在所述化学式1中,r1及r3独立地为氢原子、c1‑
c
10
烷基或烷氧基,r2为下述化学式2的取代基,n为2以上的数。
[0028]
[化学式2]
[0029][0030]
在化学式2中,r4及r5独立地为氢原子、c1‑
c
10
烷基或烷氧基,r6为c1‑
c
10
伸烷基或单共价键,*表示连接部位。
[0031]
根据一实例,可包括以下步骤:使用酸与碱对在所述(c)步骤中得到的无机颗粒进行处理,以得到表面电荷得到控制的无机颗粒。
[0032]
根据一实例,所述溶剂可为水、或与水具有兼容性的溶剂和水的混合溶剂。
[0033]
根据一实例,所述与水具有兼容性的溶剂可为选自醇、氯仿、乙二醇、丙二醇、二乙二醇、甘油及丁二醇中的一种以上。
[0034]
另外,根据本发明,提供一种水分散液,在水中分散有所述的无机颗粒。
[0035]
根据一实例,所述水分散液可为用于cmp的浆料。
[0036]
发明效果
[0037]
本发明的无机颗粒是通过将结晶相与非晶相混合且结晶度为90%以下的多个小颗粒凝聚而形成的无机颗粒,且所述小颗粒具有形成表面凸块的形状,因此可具有大的比表面积,且易于通过ph调节来控制表面电荷。因此,不仅增大了与硅膜的接触面积,还提高了研磨速度,且划痕损伤少,因此在用作cmp研磨浆料中包含的研磨颗粒时,研磨效率优异。
附图说明
[0038]
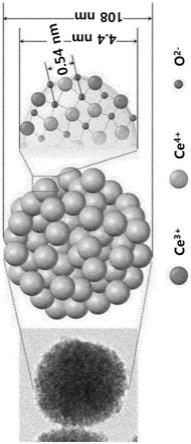
图1是概略性示出根据本发明的无机颗粒的形状。
[0039]
图2是关于实施例1及比较例1的ceo2颗粒的扫描电子显微镜(scanning electron microscope)影像与高分辨率透射电子显微镜(high resolution transmission electron microscope,hr
‑
tem)影像。
[0040]
图3是根据比较例2的样品的扫描电子显微镜(scanning electron microscope)影像。
[0041]
图4是对实施例1及比较例1的ceo2颗粒的粒度分布进行分析的直方图。
[0042]
图5是关于实施例1及比较例1的ceo2颗粒的x射线衍射(x
‑
ray diffraction,xrd)分析结果。
[0043]
图6表示根据实施例1及比较例1的ceo2颗粒的hr
‑
tem影像与选择区域电子衍射(selected area electron diffraction,saed)图案。
[0044]
图7是根据实施例1及比较例1的ceo2颗粒的x射线光电子能谱(x
‑
ray photoelectron spectroscopy,xps)分析结果。
[0045]
图8是关于实施例1及比较例1的ceo2颗粒的水分散液的ζ电位(zeta potential)测量结果。
[0046]
图9是比较使用根据实施例1及比较例1的ceo2颗粒的浆料的硅膜的研磨率(移除速率(removal rate))的结果。
[0047]
图10是分别使用实施例1的ceo2颗粒(图10的(a))与比较例1的颗粒(图10的(b))
进行cmp测试(test)之后的对晶圆表面的原子力显微镜(atomic force microscope)影像。
具体实施方式
[0048]
以下,将参考各种实例对本发明更详细地进行说明。
[0049]
然而,这并不旨在将本发明限定为特定的实施形态,应理解为包括本发明的技术思想及范围内包括的所有变形物、等同物或替代物。
[0050]
可使用第一、第二、a、b等用语术语来说明各种构成要素,但是所述构成要素不受所述用语的限制,且仅出于将一个构成要素与其他构成要素区分开的目的而使用。
[0051]“及/或”的用语包括多个记载的项目中的任一者或包括其等的组合。
[0052]
在将某一构成要素称为“连接”或“相接”至另一构成要素时,应理解为它直接连接或相接至另一构成要素,或者中间还可能存在其他构成要素。
[0053]
除非另有说明,否则单数表现包括复数表现。
[0054]“具备”、“包含”或“具有”等用语是指说明书中记载的特征、数值、步骤、动作、构成要素、零件或其组合的存在,且并不排除可能存在或添加未提及的其他特征、数值、步骤、动作、构成要素、零件或其组合的可能性。
[0055]
根据本发明,通过使自组装界面活性剂在水系溶剂中与无机物前驱体反应,可合成并非根据无机物固有的原子组装特性的颗粒形状的其他形状。例如,可将根据固有的原子组装结构只能形成呈现出角的萤石(fluorite)六边形结构的二氧化铈(ceo2)无机颗粒制备成具有凸块的球状颗粒。
[0056]
根据本发明,混合有结晶相与非晶相且结晶度为90%以下的多个小颗粒凝聚形成无机颗粒。所述结晶度也是小颗粒的结晶度,但是由于小颗粒凝聚形成纳米簇(nanocluster)形态的无机颗粒,因此也可称为无机颗粒的结晶度。所述结晶度还表示整体相中结晶相所占的比率。即,结晶度为90%以下是指结晶相为90%以下而非晶相占有10%以上。所述小颗粒或无机颗粒的结晶度可为90%以下、85%以下、80%以下或75%以下,且可为50%以上、60%以上、65%以上、或70%以上。
[0057]
由于结晶相与非晶相以一定比率混合,因此在用作cmp工程的研磨浆料时,可将基板的划痕、凹陷(dishing)等缺陷的产生最小化。
[0058]
图1概略性示出根据本发明的无机颗粒的结构。即,根据本发明的无机颗粒由非常小的小颗粒的集合体形成,且由于小颗粒混合有结晶相与非晶相并形成表面凸块,因此具有非常独特的表面。
[0059]
如图1所示,无机颗粒及小颗粒实质上均为球状。在此,球状是指以短径/长径之比表示的纵横比(aspect ratio)为0.8以上、0.9以上、或0.95以上,且为1.2以下、1.1以下或1.05以下。因此,当提及根据本发明的无机颗粒时,在下文中还称为“球状凸块无机颗粒”或“球状凸块颗粒”。
[0060]
由于无机颗粒在其表面具有球状凸块,因此具有可基于相同质量增加颗粒的比表面积的效果。形成球状凸块的小颗粒的直径为无机颗粒直径的2%至25%。优选可为5%以上、10%以上、15%以上或20%以上。小颗粒的粒径可为10nm以下、8nm以下、6nm以下、或5nm以下,且可为1nm以上、2nm以上、3nm以上或4nm以上。
[0061]
根据本发明的球状凸块无机颗粒具有30nm至1000nm的粒度分布且形成为均匀的
大小。球状凸块无机颗粒的大小基于数平均粒径优选为50nm以上、100nm以上、110nm以上、或120nm以上且800nm以下、500nm以下、300nm以下、200nm以下或150nm以下。无机颗粒粒径的标准偏差可为20以下、18以下、16以下、14以下、12以下或11以下。
[0062]
图1概略性示出根据本发明实施例1制备的二氧化铈颗粒的结构,且示出ceo2单位体(大小为0.54nm)被聚集以形成小颗粒(粒径为4.4nm),且小颗粒被聚集以形成二氧化铈颗粒(粒径为108nm)。
[0063]
根据本发明的球状凸块无机颗粒可通过自组装界面活性剂与无机物前驱体进行自组装反应来制备,因此根据本发明的无机颗粒其密度为3.0g/ml至5.0g/ml。密度可通过tap密度测量法(astm b527)来测量。无机颗粒的密度可为3.2g/ml以上、3.3g/ml以上、3.4g/ml以上或3.5g/ml以上,且可为4.5g/ml以下或4.0g/ml以下。
[0064]
根据一实例,所述一次颗粒及二次颗粒可分别独立地为由选自由以下所组成的群组中的一种以上的元素的氧化物形成:ga、sn、as、sb、ce、si、al、co、fe、li、mn、ba、ti、sr、v、zn、la、hf、ni及zr。根据优选的实施例,可为选自铈(ce)、硅(si)及铝(al)的一种以上的氧化物。根据优选的实施例,无机颗粒可由二氧化铈(ceo2)形成。
[0065]
根据一实例,所述球状凸块无机颗粒在水分散液状态下可至少具有一次+30mv以上、或
‑
30mv以下的表面电荷,特别是在ph4的条件下表现出+30mv至+50mv或
‑
30mv至
‑
50mv的绝对值高的表面电荷(ζ电位)。在此,“表面电荷”的用语作为与“ζ电位”相同的含义使用。
[0066]
另外,根据本发明的实施例,无机颗粒的等电点可为ph 5~ph 7。优选为等电点可为5.5以上且6.5以下的ph。在水系中,等电点越低表示在颗粒表面存在的oh
‑
基越多,这即意味着在颗粒表面的活性部位多,因此对于在cmp工程中提高研磨性能方面是有利的。
[0067]
本发明还提供一种将上述无机颗粒分散在水中的水分散液。
[0068]
在将根据本发明的无机颗粒在半导体cmp工程中用作浆料内的研磨颗粒时,由于是不存在锐利的角的球状且包含非晶相,因此可补偿划痕不良缺陷,且由于在颗粒表面存在许多凸块,因此比表面积增加,从而不仅提高与待研磨的膜质接触的概率,而且由于颗粒表面性质的改变而可提高研磨速度。例如,在通过本发明提出的方法制备的球状凸块二氧化铈颗粒的情况下,由于颗粒表面上的元素缺陷,与现有的六边形萤石(fluorite)二氧化铈颗粒相比,ce(iii)变多,从而可提高研磨速度。
[0069]
根据本发明制备的二氧化铈颗粒的ce
3+
/ce
4+
离子比可为40至60。ce
3+
/ce
4+
离子比越高,研磨速度越高,根据本发明可得到40以上、42以上、44以上、或46以上的离子比。所述离子比可为60以下、55以下或50以下。
[0070]
另外,通过使用本发明提出的通过ph调节来控制无机颗粒的表面电荷的方法,可更容易地控制球状凸块无机颗粒的表面电荷,且通过使用该方法营造在cmp工程中可发挥研磨颗粒与膜质间的最佳相互作用的水溶液的ph环境,由此可更有效且稳定地进行研磨。
[0071]
在下文中,将对根据本发明的使用液相合成法制备球状凸块无机颗粒的方法更具体地进行说明。
[0072]
使用液相合成法制备球状凸块无机颗粒的方法
[0073]
根据本发明的球状凸块无机颗粒可通过包括下述步骤的方法来制备。
[0074]
(a)使自组装界面活性剂溶解于溶剂的步骤;
[0075]
(b)在实施所述(a)步骤之前、之后或同时,使无机物前驱体溶解或分散于所述溶
剂中来制备无机物前驱体溶液的步骤;以及
[0076]
(c)通过所述无机物前驱体与所述界面活性剂的自组装反应,在界面活性剂形成的壳中形成混合有结晶相与非晶相的小颗粒,进而多个小颗粒凝聚形成无机颗粒的步骤。
[0077]
在使用本发明中提出的液相合成法制备球状凸块无机颗粒的过程中,步骤(c)的颗粒形成过程包括以下步骤:(i)在将无机物前驱体与自组装界面活性剂一起还原的同时形成小颗粒;以及(ii)进行自组装界面活性剂的自组装反应的同时,在多个小颗粒凝聚时生长为在表面具有凸块的球状无机颗粒。尽管分开说明了形成无机颗粒与形成表面凸块的两个步骤,但是由于反应是连续进行的,因此可看作通过一个合成步骤形成球状凸块无机颗粒。
[0078]
无机物前驱体
[0079]
首先,制造要制备的无机物的前驱体溶液。通过将无机物前驱体与自组装界面活性剂、溶剂混合来制备,此时可在将界面活性剂首先溶解于溶剂中之后添加无机物前驱体,且还可首先将无机物前驱体溶解于溶剂中之后添加界面活性剂并混合,或者还可在溶剂中同时添加无机物前驱体与自组装界面活性剂并混合。在此过程中,在无机物前驱体与界面活性剂之间形成了弱键。
[0080]
在此,作为无机物前驱体,是包含选自由ga、sn、as、sb、ce、si、al、co、fe、li、mn、ba、ti、sr、v、zn、la、hf、ni及zr组成的群组中的一种以上的元素且可形成氧化物的物质。在本发明中使用的无机物前驱体优选为在水溶液状态下可与带电荷的界面活性剂离子键合的化合物形态。例如,可为硝酸盐、溴化物、碳酸盐、氯化物、氟化物、氢氧化物、碘化物、草酸盐或硫酸盐,且其等可为水合物或无水物形态。
[0081]
更具体而言,可使用如下等包含铈的盐:例如,硝酸铈(iv)铵(ammoniumcerium(iv)nitarate)、无水溴化铈(iii)(cerium(iii)bromide anhydrous)、水合碳酸铈(iii)(cerium(iii)carbonate hydrate)、无水氯化铈(iii)(cerium(iii)chloride anhydrous)、七水合氯化铈(iii)(cerium(iii)chloride heptahydrate)、无水氟化铈(iii)(cerium(iii)fluoride anhydrous)、氟化铈(iv)(cerium(iv)fluoride)、氢氧化铈(iv)(cerium(iv)hydroxide)、无水碘化铈(iii)(cerium(iii)iodide anhydrous)、六水合硝酸铈(iii)(cerium(iii)nitrate hexahydrate)、水合草酸铈(iii)(cerium(iii)oxalate hydrate)、硫酸铈(iii)(cerium(iii)sulfate)、水合硫酸铈(iii)(cerium(iii)sulfate hydrate)、八水合硫酸铈(iii)(cerium(iii)sulfate octahydrate)、水合硫酸铈(iv)(cerium(iv)sulfate hydrate)。
[0082]
作为另一例,可使用如原硅酸四乙酯(tetraethyl orthosilicate,teos)、二乙氧基二甲基硅烷(diethoxydimethylsilane,dems)及乙烯基三乙氧基硅烷(vinyltriethoxysilane,vtes)等硅前驱体、具有ti(or)4结构的钛前驱体、具有zr(or)4结构的锆前驱体、具有al(or)4结构的铝前驱体等。在此,r是指可与水或醇水合化或醇化的官能基,例如可为如甲基、乙基等低级烷基。除此之外,还可使用可形成ga、sn、as、sb、mn或v的氧化物的前驱体。
[0083]
自组装界面活性剂
[0084]
作为形成自组装的界面活性剂,阴离子性界面活性剂、阳离子性界面活性剂及两性界面活性剂均可使用,且可与无机物前驱体结合且在溶解于溶剂时具有(+)或(
‑
)或同时
具有两种电荷,且保有可通过交联反应引导颗粒形成反应的官能基。作为这种官能基,可列举酰胺基、硝基、醛基、羰基等。
[0085]
根据本发明,可根据合成反应中使用的自组装界面活性剂的种类来制备表面电荷不同的颗粒。即,可根据要制备的无机颗粒的表面电荷选择性地使用自组装界面活性剂。例如,在要制备带有(
‑
)电荷的球状凸块无机颗粒的情况下可使用阳离子性界面活性剂。通过在阳离子性界面活性剂的带有(+)电荷的部分中与无机物前驱体的离子键合而形成小颗粒,随着反应的进行,形成了自组装的壳且其中无机颗粒生长为在表面具有凸块的球状形状。根据相同的原理,相反地,在要制备具有(+)电荷的球状凸块无机颗粒的情况下,可使用阴离子性界面活性剂。如上所述,为了制备具有目标表面电荷的无机颗粒,需要具有特定离子性的界面活性剂壳,且可根据所使用的自组装界面活性剂的种类来制备表面电荷不同的颗粒。
[0086]
另外,根据需要,可在合成过程中将一种或一种以上的界面活性剂混合使用。在自组装物质中,界面活性剂可在溶解于溶剂的同时彼此形成交联(crosslinking),并在一定温度与一定时间以上随着反应的进行而自组装。此时,与该界面活性剂结合的小颗粒之间的间隔变近聚集并使颗粒生长,由自组装的界面活性剂的壳包围生长而形成其中充满球状颗粒的无机颗粒,同时生长为在表面包含许多凸块的形状。凸块可在球状颗粒的表面上同时生长,独立生长的凸块还可在球状颗粒的表面出现并形成凸块。
[0087]
作为阴离子性界面活性剂,可使用烷基苯磺酸盐(alkylbenzene sulfonates)、烷基硫酸盐(alkyl sulfates)、烷基醚硫酸盐(alkyl ether sulfates)、肥皂(soaps)等。
[0088]
作为阳离子性界面活性剂,可使用烷基季氮(alkyl quaternary nitrogen)化合物、如酯季铵盐(esterquats)等季铵(quaternary ammonium)化合物等。
[0089]
另外,可使用既包含阳离子性的季铵离子(quaternary ammonium ion)基还包含阴离子性的羧酸盐(
‑
coo
‑
)、硫酸盐(
‑
so42
‑
)或磺酸盐(
‑
so3
‑
)基的两性界面活性剂。
[0090]
此外,吡啶甲酸(picolinic acid)、(羧甲基)二甲基
‑3‑
[(1
‑
氧十二烷基)胺基]丙基氢氧化铵((carboxymethyl)dimethyl
‑3‑
[(1
‑
oxododecyl)amino]propylammonium hydroxide)、月桂基甜菜碱(lauryl betaine)、甜菜碱柠檬酸盐(betaine citrate)、月桂酰两性基乙酸钠(sodium lauroamphoacetate)、羟甲基甘胺酸钠(sodium hydroxymethylglycinate)、(羧甲基)二甲基油铵氢氧化物((carboxymethyl)dimethyloleylammonium hydroxide)、椰油酰胺基丙基甜菜碱(cocamidopropyl betaine)、(羧甲基甲基)二甲基(十八烷基)铵((carboxylate methyl)dimethyl(octadecyl)ammonium)、聚环氧乙烷
‑
聚环氧丙烷嵌段共聚物(peo
‑
ppo block copolymer)、阴离子硅氧烷(anionic siloxane)及树枝状聚合物(dendrimers)、聚(10
‑
十一碳烯酸钠)(poly(sodium 10
‑
undecylenate))、聚(10
‑
十一烯基硫酸钠)(poly(sodium 10
‑
undecenylsulfate))、聚(十一烯酰基缬胺酸钠)(poly(sodium undeconylvalinate))、聚乙烯吡咯啶酮(polyvinylpyrrolidone)、聚乙烯醇(polyvinylalcohol)、2
‑
丙烯酰胺
‑2‑
甲基
‑1‑
丙磺酸(2
‑
acrylamide
‑2‑
methyl
‑1‑
propanesulfonic acid)、甲基丙烯酰胺烷基酯(alkyl methacrylamide)、丙烯烷基酯(alkyl acrylate)、支撑聚(烯丙基胺)的相(poly(allylamine)
‑
supported phase)、聚(次乙亚胺)(poly(ethyleneimine))、聚(n
‑
异丙基丙烯酰胺)(poly(n
‑
isopropylacrylamide))、n
‑
羟基琥珀酰亚胺(n
‑
hydroxysuccinimide)
等。
[0091]
优选地,所述自组装界面活性剂可为下述化学式1的高分子。另外,下述化学式1的高分子可以是分子内(+)与(
‑
)性质均具有的两性界面活性剂。
[0092]
[化学式1]
[0093][0094]
在所述化学式1中,r1及r3独立地为氢原子、c1‑
c
10
烷基或烷氧基,n为2以上的数,r2为具有下述化学式2的结构的取代基。
[0095]
[化学式2]
[0096][0097]
在化学式2中,r4及r5独立地为氢原子、c1‑
c
10
烷基或烷氧基,r6为c1‑
c
10
伸烷基或单共价键,*表示连接部位。
[0098]
所述化学式1的高分子的分子量优选为500以上且100,000以下。在此,分子量是重量平均分子量,且重量平均分子量是指通过凝胶渗透色谱法(gel permeation chromatography,gpc)测量的以聚苯乙烯换算的分子量。所述分子量可为1000以上、5000以上、10,000以上、20,000以上或30,000以上,且可为95,000以下、90,000以下、85,000以下、80,000以下、70,000以下、60,000以下、50,000以下或40,000以下。
[0099]
对于每100重量份无机物前驱体,自组装界面活性剂的使用量可为30重量份至150重量份。对于每100重量份无机物前驱体,界面活性剂的使用量可为40重量份以上、50重量份以上、60重量份以上、70重量份以上、80重量份以上或90重量份以上,且可为140重量份以下、130重量份以下、120重量份以下或110重量份以下。
[0100]
溶剂
[0101]
球状凸块无机颗粒的合成反应中使用的溶剂可为水、或与水具有兼容性的溶剂和水的混合溶剂。
[0102]
根据一实例,所述与水具有兼容性的溶剂可为选自醇、氯仿、乙二醇、丙二醇、二乙二醇、甘油及丁二醇中的一种以上。
[0103]
在将与水具有兼容性的溶剂和水混合使用的情况下,水:兼容性溶剂的混合体积比可为100:50至200、或100:60至150、或100:70至120。
[0104]
在使用水或与水具有兼容性的溶剂和水的混合物作为溶剂并添加无机物前驱体及/或自组装界面活性剂并使其溶解时,宜使用搅拌器,且宜在完全溶解后进行反应。否则,可能会妨碍形成均匀形态(morphology)的颗粒。
[0105]
球状凸块无机颗粒的合成反应
[0106]
在球状凸块无机颗粒的合成步骤中,将前文制备的无机物前驱体溶液引入反应器
并与自组装界面活性剂进行合成反应。球状凸块无机颗粒的合成在60℃至250℃的温度范围内进行1小时至24小时。优选为2小时以上、3小时以上、或4小时以上,且在20小时以下、10小时以下或8小时以下,可在70℃以上、80℃以上或90℃以上且在220℃以下、200℃以下、180℃以下或160℃以下的范围内进行。
[0107]
将自组装界面活性剂溶解于溶剂中,然后随着反应在一定温度与时间内进行而与无机物前驱体的离子结合。在此,自组装是指界面活性剂的具有(+)性质的部分与具有(
‑
)性质的部分在结合时自发地形成组织结构或形态。例如,若界面活性剂在分子结构内具有酰胺基,则氮原子部分具有(+)性质,而氧原子部分具有(
‑
)性质,从而可自然地形成网络结构。同时,与这种自组装物质一起溶解于溶剂中的小颗粒之间的间隔变近并凝聚,同时颗粒生长(形成纳米簇)。在此过程中,颗粒被界面活性剂壳包围并生长,因此制成球状颗粒,且在颗粒表面形成凸块。此时,凸块可在球状颗粒的表面上同时生长,或者独立生长的凸块可在球状颗粒的表面出现并形成凸块。
[0108]
球状凸块无机颗粒的表面电荷控制方法
[0109]
根据本发明,使用酸及/或碱对所述合成反应中得到的无机颗粒进行处理以控制无机颗粒的表面电荷。
[0110]
本发明中提出的控制球状凸块无机颗粒的表面电荷的方法实质是控制包含颗粒的水溶液的ph。例如,若水溶液内存在带正电的颗粒,则越添加酸性物质颗粒渐渐带有更强的正电荷;相反,越添加碱性物质颗粒的表面电荷逐渐带有弱的正电荷并达到中性。若继续添加过多的碱,则将带有负电荷。可通过利用这种原理调节水溶液中的ph来控制无机颗粒的表面电荷。
[0111]
作为用于降低水溶液ph的酸性ph调节剂,可使用一种或混合使用一种以上酸性物质,例如磷酸、盐酸、硝酸、硫酸等,相反,作为用于提高ph的碱性ph调节剂,可使用一种或混合使用一种以上碱性物质,例如氢氧化钠、氨水等。此时,仅在调节ph的同时使用搅拌器将水溶液内部均匀混合时,才可实现准确的ph测量。
[0112]
根据本发明的球状凸块无机颗粒是至少具有一次表面电荷为+30mv以上或
‑
30mv以下的无机颗粒,包括以稳定状态存在于水溶液内由此可更有效地发挥表面特性的表面电荷调节方法。如此制备的颗粒与如玻璃、硅等各种介质的结合力优异,因此可用作研磨颗粒。
[0113]
特别是根据本发明的无机颗粒在ph4的水分散液状态下表面电荷可为+30mv至+50mv或
‑
30mv至
‑
50mv。即,由于在给定的ph条件下具有绝对值高的ζ电位,因此研磨速率将进一步得到提高。在此,“表面电荷”的用语作为与“ζ电位”相同的含义使用。
[0114]
用于实施发明的形态
[0115]
通过以下实施例将更详细地说明本发明的构成及作用。然而,此仅作为本发明的优选示例而提出,并在任何意义上均不能由此解释为对本发明的限制。另外,将省略本技术领域的技术人员可在技术上充分推导出的内容的说明。
[0116]
球状凸块二氧化铈颗粒的制备
[0117]
<实施例1>
[0118]
在以100:100的体积比混合乙二醇(99%)与水的160ml溶剂中,添加2g聚(n
‑
异丙基丙烯酰胺)(poly(n
‑
isopropylacrylamide))(奥德里奇(aldrich)公司,mw:30,000)作为
自组装界面活性剂并使用磁搅拌器搅拌。确认完全溶解后,添加2g奥德里奇公司(aldrich corporation)的硝酸铈六水合物(cerium nitrate hexahydrate)(ce(no3)3·
6h2o)作为铈前驱体并溶解以制备铈前驱体溶液。
[0119]
将该铈前驱体溶液添加至保持温度的液相反应器中,在90℃至140℃的温度范围内进行约165分钟的合成反应。反应完成后,使用离心分离器在4000rpm下将得到的二氧化铈颗粒溶液离心分离1小时30分钟,分离出沉淀物,之后重复3次用水(h2o)洗涤的过程,以得到作为结果物的二氧化铈颗粒(以下还称为“boc100”)。
[0120]
<实施例2>
[0121]
在180ml水中溶解有2.4g氯化铈(cerium chloride)的水溶液中添加与实施例1中使用者分子量不同的2.4g聚(n
‑
异丙基丙烯酰胺)(poly(n
‑
isopropylacrylamide))(奥德里奇(aldrich)公司,mw:85,000),并在70℃至90℃下搅拌6小时进行反应。之后,使用与上述实施例1相同的方法进行分离及洗涤,得到具有球状凸块的二氧化铈颗粒。
[0122]
<比较例1>
[0123]
准备萤石(fluorite)六边形结构的ceo2颗粒(制造商:索尔维(solvay),产品名:hc60)。
[0124]
<比较例2>
[0125]
将8g奥德里奇公司(aldrich corporation)的硝酸铈六水合物(cerium nitrate hexahydrate)(ce(no3)3·
6h2o)作为铈前驱体添加至160ml水中使其溶解,制成铈前驱体溶液并使用磁搅拌器进行搅拌。确认完全溶解后,添加4g氢氧化钠(sodium hydroxide)(naoh)以制备碱性状态的溶液。搅拌约1小时来准备通过沉淀法(precipitation method)合成的ceo2颗粒。
[0126]
形态及结构分析
[0127]
使用扫描电子显微镜(fe
‑
sem,jeol jsm 7401f)、高分辨率透射电子显微镜(hr
‑
tem,jem
‑
2100f)、x射线衍射分析仪(具有cu kα辐射的日本理学(rigaku)smartlab se x射线衍射仪(x
‑
ray diffractometer))及x射线光电子能谱仪(xps,赛默飞(thermo)escalab 250)对实施例1及实施例2与比较例1的二氧化铈颗粒的形态及结构进行分析。
[0128]
图2的a及图2的b是示出在实施例1中制备的二氧化铈颗粒(boc100)的形状的sem影像与tem影像。表明实施例1的二氧化铈颗粒是圆形的且具有平滑表面的球形状。
[0129]
另一方面,根据图2的d及图2的e,示出比较例1的二氧化铈颗粒(hc60)保有尖锐角的隅角与晶格表面的萤石晶体的特征性形状。
[0130]
图3是根据比较例2藉由沉淀法制备的颗粒的sem影像。可确认颗粒的形状是不规则且结块的形状。
[0131]
另外,图4是表示实施例1及比较例1的二氧化铈颗粒的粒度分布的直方图。实施例1的二氧化铈颗粒(boc100)的平均粒径为108nm,标准偏差为10.3,但比较例1的二氧化铈颗粒(hc60)的平均粒径为117nm,标准偏差为22.5。根据实施例1的二氧化铈颗粒(boc100)的标准偏差较比较例1的二氧化铈颗粒(hc60)的标准偏差小得多的事实,可确认实施例1的二氧化铈颗粒表现出单分散性。
[0132]
具有球形状且具有单分散性的根据本发明的二氧化铈颗粒由于没有呈现出角的隅角且具有圆的且缓的表面,因此在减少cmp工程中的缺陷、划痕或凹陷瑕疵方面是更佳
的。
[0133]
如图1所示,根据实施例1的二氧化铈颗粒由非常小的微细纳米颗粒(约4.4nm的粒径)形成,因此具有非常独特的表面。即,根据本发明的无机颗粒形成为纳米颗粒或单位体的聚合体,在实施例1的情况下,ceo2原子(0.54nm)被聚集以形成纳米颗粒(4.4nm),且可以说纳米颗粒被聚集形成无机颗粒(108nm)。
[0134]
图5是对实施例1与比较例1的二氧化铈颗粒的xrd分析结果。两个颗粒均在与对应于萤石晶体的特征性波峰的(111)、(200)、(220)、(311)、(222)、(400)、(331)及(420)晶格面相应的28.55
°
、33.08
°
、47.47
°
、56.33
°
、59.08
°
、69.4
°
、76.7
°
及79.07
°
处具有波峰。然而,由于实施例1的二氧化铈颗粒(boc100)的波峰宽得多,因此可知其结晶度低于比较例1的hc60颗粒的结晶度。
[0135]
另外,为比较结晶大小,基于(111)波峰计算结晶大小。通过使用半峰全幅值(full
‑
width
‑
of
‑
half
‑
maximum,fwhm)的谢乐方程式(scherrer equation)计算平均结晶大小(lc)(莫什,a.(monshi,a.),m.r.福鲁吉(m.r.foroughi)与m.r.莫什(m.r.monshi),修改后的谢乐方程式以使用xrd更准确地估算纳米晶的尺寸(modified scherrer equation to estimate more accurately nano
‑
crystallite size using xrd),世界纳米科学与工程杂志(world journal of nano science and engineering),2012.02(03):p.154
‑
160)。
[0136]
[数学式1]
[0137][0138]
在所述式中,λ是x射线波长(nm),β是fwhm(弧度),且k是与晶体形状有关的常数(0.9)。
[0139]
使用ruland
‑
vonk方法利用在xrd波峰下的面积计算颗粒的结晶度(尤莲奈丽(iulianelli),g.c.v.等人,tio2纳米颗粒对phb基质的热学、形态及分子特性的影响(influence of tio2 nanoparticle on the thermal,morphological and molecular characteristics of phb matrix),聚合物测试(polymer testing),2018,65:p.156
‑
162)。
[0140]
[数学式2]
[0141][0142]
在所述式中,ic是结晶波峰下面积的和,ia是非晶态分割面积的和。
[0143]
如表1中整理所示,实施例1的二氧化铈颗粒的晶体大小(lc)为4.4nm,结晶度xc为70.5%。在图1的概略图中,4.4nm的纳米颗粒大小基于这种xrd分析的结果。
[0144]
[表1]
[0145]
[0146]
另一方面,比较例1的颗粒的结晶大小(lc)为45.5nm,且结晶度(xc)为95.8%。即,根据本发明的实施例1的颗粒的结晶大小较比较例1的颗粒的晶体大小小得多,且结晶度低得多。由此,可知根据本发明的颗粒包含大量的非晶二氧化铈。非晶相相比结晶相软得多,因此较佳为用于减轻cmp工程中的划痕或凹陷缺陷。
[0147]
图6的b与图6的c示出根据实施例1的颗粒的hr
‑
tem影像与选择性区域电子衍射(saed)图案。实施例1的颗粒的d
‑
间距(d
‑
spacing)为其对应于二氧化铈的(111)晶格面。在图6的c中,绘制相的边界而得到的相的大小在2nm至5nm的范围内。此对应于图1中记载的4.4nm的晶体大小。扩散的saed图显示在图6的d中,表明实施例1的颗粒是结晶相及非晶相的混合物(由点与环表示)。
[0148]
图6的e至图6的g示出比较例1的颗粒在被边界线包围的一个方向上具有由(111)晶格面表示的大结晶相。此表明比较例1的颗粒由几乎一个或两个单晶相形成,其由45.5nm的晶体大小与95.8%的结晶度支撑。
[0149]
图7分别由曲线图a与曲线图b示出使用x射线光电子能谱分析(xps)对实施例1与比较例1的颗粒针对ce 3d与o1s实施元素分析的结果。图7的a示出ce 3d波峰分成ce 3d
5/2
与ce 3d
3/2
。
v0
、
v1
、
v2
、
v3
及
v4
属于ce 3d
5/2
,而
u0
、
u1
、
u2
、
u3
及
u4
属于ce 3d
3/2
(索麦特,n.(thromat,n.),m.戈蒂埃
‑
索亚(m.gautier
‑
soyer)与g.波迪尔(g.bordier),形成cey2o3界面:原位xps研究(formation of the cey2o3 interfave:an in situ xps study),表面科学(surface science),1996.345(3):p.290
‑
302)。
v0
、
v2
、
u0
及
u2
波峰表示ce
3+
离子的特性,
v1
、
v3
、
v4
、
u1
、
u3
及
u4
波峰表示ce
4+
离子的特性(张,c(zhang,c.)与j.林(j.lin),可见光诱导的四方zro2
–
ceo2固溶体中的氧桥式zr iv
‑
o
‑
ce iii氧化还原中心,用于降解有机污染物(visible
‑
light induced oxo
‑
bridged zr iv
‑
o
‑
ce iii redox centre in tetragonal zro2
–
ceo2 solid solution for degradation of organicpollutants),物理化学化学物理(physical chemistry physics),2011.13(9):p.3896
‑
3905)。
[0150]
ce
3+
与ce
4+
的浓度如下所示般求得。
[0151]
[ce
3+
]=
v0
+
v2
+
u0
+
u2
[0152]
[ce
4+
]=
v1
+
v3
+
v4
+
u1
+
u3
+
u4
[0153]
表2表示xps波峰分配的具体信息。
[0154]
[表2]
[0155][0156]
根据所述结果,具有实施例1的颗粒的ce
3+
计算浓度为32.6%,高于比较例1的颗粒的28.3%。另外,对于ce
3+
/ce
4+
的比率,实施例1的颗粒为48.4,比较例1的颗粒为39.5,可知实施例1的颗粒中所含的ce
3+
的浓度更高。
[0157]
在水系中,存在于二氧化铈颗粒表面的ce
3+
离子会促进h2o的解离,从而在ceo2表面形成羟基(oh group)。颗粒表面的羟基不仅起到活性点的作用,而且还有助于其他物质
的物理吸附,特别是在cmp工程中形成ce
‑
o
‑
si键。
[0158]
对实施例1及比较例1的颗粒通过o1s xps分析法来测量羟基浓度(参见图7的b)。528.83ev的波峰对应于晶格氧离子o2‑
,而530.33ev的波峰对应于表面氢氧根离子oh
‑
(参见表2)(范登布兰德(van den brand,j.)等人,由氧化铝层的xps光谱确定的羟基分数与o/al原子比率之间的相关性(correlation between hydroxyl fraction and o/al atomic ratio as determined from xps spectra of aluminium oxide layers)。表面与界面分析:国际期刊,致力于表面、界面与薄膜分析技术的开发和应用(surface and interface analysis:an international journal devoted to the development and application of techniques for the analysis of surfaces,interfaces and thin films),2004.36(1):p.81
‑
88)。
[0159]
实施例1的颗粒在表面具有69.4%的oh
‑
,此为较比较例1的颗粒具有的47.3%的oh
‑
高得多的量。另外,实施例1的颗粒在表面具有30.6%的o2‑
,其为较比较例1的颗粒具有52.7%的o2‑
低得多的浓度。这种结果与实施例1的颗粒表面具有较比较例1的颗粒高得多的ce
3+
离子浓度相应。因此,可以说表面的oh
‑
浓度与存在的ce
3+
的量成比例。结果,具有较比较例1的颗粒更多的ce
‑
oh活性部位的实施例1的颗粒可促进在cmp工程中在sio2基板与ceo2颗粒之间形成ce
‑
o
‑
si键。
[0160]
密度
[0161]
通过tap密度测量法(astm b527)测量根据实施例1及实施例2以及比较例1及比较例2的ceo2无机颗粒的密度。
[0162]
[表3]
[0163][0164]
ζ电位测量
[0165]
使用马尔文(malvern)公司的ζ电位分析仪(ζpotential analyzer)(nano zs)测量ζ电位。
[0166]
图8是使用硝酸溶液(酸性ph调节剂)与氨水(碱性ph调节剂)将制备例1的球状凸块ceo2颗粒分散液的ph调节为ph2
‑
ph10后测量ζ电位(zeta potential)的结果。
[0167]
如图8所示,实施例1及比较例1的浆料在ph2时显示出约60mv的高正电荷,然后随着ph值的增加而显示出弱的正电荷。在对应于cmp工程条件的ph为4~4.5时,两种浆料的ζ电位均大于30mv,并通过静电排斥力而保持稳定分散的状态。另一方面,由于二氧化硅颗粒的ζ电位在ph为2至10的宽范围内,特别是在ph为4附近为负,因此二氧化硅基板与二氧化铈颗粒之间由于彼此相反电荷而产生静电吸引。在去离子水中,实施例1的颗粒的等电点(iep)大致在ph6附近,而比较例1的颗粒在ph9附近。判断实施例1的颗粒的oh
‑
浓度较高,其
原因如在xps分析中所见。
[0168]
研磨性能测试
[0169]
制备将实施例1及比较例1的二氧化铈颗粒分别以0.1重量%、0.2重量%、0.3重量%、0.4重量%、0.5重量%、0.6重量%、0.7重量%、0.8重量%、0.9重量%、1.0重量%、2.0重量%及3重量%的浓度分散在去离子水中的浆料,而不存在其他添加剂。
[0170]
使用gnp poli
‑
400l在浆液流速(flow rate):150ml/min、固定头压力(fixed head pressure):4psi的条件下进行cmp测试(cmp test)1分钟。裸晶圆(bare wafer)上的sio2初始厚度为并使用折光仪(st4000
‑
dlx)测量移除速率(removal rate,rr)(参见图9)。
[0171]
已知包含比较例1的颗粒的浆料通常在0.3重量%的浓度时表现出最高的研磨性能。因此,比较颗粒浓度为0.3重量%时的rr的结果,实施例1测定为能。因此,比较颗粒浓度为0.3重量%时的rr的结果,实施例1测定为比较例1测定为
[0172]
另外,比较例1的颗粒的rr逐渐增加直至浓度增加至2重量%时为止,然后在3重量%减少。反之,实施例1的颗粒的rr随着其浓度的增加而增加,且在3重量%下达到的rr。此为在比较例1的颗粒在2重量%下达到的rr、即达到的233%的高的值。由此可知,比较例1的颗粒在约2重量%下饱和,但是实施例1的颗粒即使至3重量%仍不饱和。这种结果被认为是由于存在于颗粒表面的ce
3+
离子与oh
‑
离子的浓度高。
[0173]
图10是通过原子力显微镜(atomic force microscope)观察使用将实施例1及比较例1的颗粒以0.3重量%分散在去离子水中的浆液进行cmp测试后的晶圆表面的模样。在利用包含比较例1的颗粒的浆料进行的cmp测试中,在晶圆表面产生深且大的划痕缺陷(图10的(b)),可通过在工程后大量的颗粒残留在表面上来确认。反之,可确认利用包含实施例1的颗粒的浆料,即使经过cmp工程在晶圆表面亦未产生划痕缺陷,残留的二氧化铈颗粒的量也非常少(图10的(a))。
[0174]
根据以上结果,可知通过本发明的方法制备的无机颗粒的大小均匀,且根据ph有效地控制表面电荷。另外,可确认能够看出通过以浆料形态进行cmp测试的结果较使用市售萤石六边形结构的二氧化铈颗粒的浆料具有优异的研磨性能,且同时在晶圆表面的划痕缺陷大大减少。
[0175]
以上,以本发明的实施例为中心进行了说明,但是对本发明所属的技术领域内具有通常知识者而言可施加各种改变或变形。这种改变与变形可在不脱离本发明提供的技术思想的范围内亦属于本发明。因此,本发明的权利范围应由所述记载的申请专利范围来判断。