1.本发明涉及液相净化技术领域,具体而言,涉及一种液相脱氯剂、其制备方法及应用。
背景技术:
2.催化重整过程中氯的来源及危害:一是在开采原油时常加入氯化物(多为有机氯化物)来提高采油率。在采油率提高的同时,也导致原油中氯含量升高。高温条件下,这部分有机氯会转化成无机氯化氢,对管路具有很大的腐蚀作用,如果下游有氨气,则会反应生成氯化铵,导致设备的堵塞,影响装置的运行。二是重整反应过程中,重整催化剂上的氯会不断流失,而且重整催化剂为了维持较高的反应活性需具备一定的酸性,所以需要不断加入水和有机氯化物,进一步导致原油中氯的含量升高,加快管路的腐蚀以及设备的堵塞。因此,制备出一种高效的脱氯剂是非常有必要的。
3.使用脱氯剂是脱除物料中氯化氢的主要方法。目前,脱氯剂主要分为两类:一类是气相脱氯剂,cn105617853a公开了一种高温气相脱氯剂,以碳酸钙、碳酸镁、氧化钙、铝酸盐水泥制成,该脱氯剂的强度低,使用周期较短,氯容有待提高。另一类是液相脱氯剂,即用于液相物料的脱氯剂,由于液相体系中传质速率较低,因此液相脱氯剂相较于气象脱氯剂的脱氯效果较差一些,cn105542836a公开了一种液相脱氯剂,由cuo、cao、koh和活性炭制成该脱氯剂的氯容只有16%。
4.如前所述,重整生成油中氯含量较低,采用液相脱氯时,脱氯剂的氯容一般较低。而且重整过程属于水氯平衡操作过程,重整生成油也含有少量的水,而水对于以碱金属为活性组分的脱氯剂来说影响较大,水会与脱氯后的物质(如氯化钙、氯化镁等)发生配合反应,造成脱氯剂结构损坏,内部孔道堵塞,最终使氯容降低。已发明的一些液相脱氯剂虽然物化参数(如比表面积、孔容、孔分布等)都比较理想,但是应用时脱氯剂的氯容仍较低,特别是以碱土金属化合物为活性组分的脱氯剂的耐水性较差。目前国内开发的液相脱氯剂的氯容质量分数控制标准在8%~15%范围内,但是根据实际情况,现有的液相脱氯剂在实际应用中的氯容均在2%以下,不能满足装置的长期运转使用,有的使用周期仅为1~2个月,更有的因为氯从脱氯剂上脱附下来导致出口氯比入口氯含量更高的情况发生。
5.氯容除了与脱氯剂本身性质相关,与操作温度也紧密相关。研究表明:对于液相脱氯,温度越高,氯容越大。而对于重整生成油液相脱氯来说,操作温度受限,一般不能超过100℃,优选不超过70℃,兰创宏等“重整生成油液相脱氯技术的设计与应用”介绍的庆阳石化公司60万t/a催化重整装置液相脱氯技术的工程设计与应用情况中,推荐液相脱氯剂在50℃操作时,氯容不足12%;在75℃操作时,氯容约为20%;在100℃操作时,氯容约为30%。同时与原料性质也有关,重整生成油中的氯含量较低,原料中含有少量水,因此现有的液相脱氯剂用于重整生成油脱氯时,一般效果较差,主要表现为氯容较低、操作周期较短等。例如福建炼厂采用的国外公司开发的液相脱氯剂,氯容只能达到3%,远远低于控制指标;uop公司开发的pcl-100分子筛吸附剂,用于液相状态下重整生成油的脱氯,寿命仅有3个月。
6.cn103830996a公开了一种高效脱氯剂。这种脱氯剂以葡萄糖和cano3、mg(no3)2、zn(no3)2、nahco3中的一种以上为活性组分混合构成,载体和助剂为zsm-5分子筛,以甲基纤维素为粘结剂,将活性组分盐滴加到经处理的载体和助剂上,然后经旋转、加热、干燥、焙烧、成型,然后造粒制得。cn1088388c公开了一种脱氯剂。该脱氯剂以钠、钙和锌的化合物为活性组分,以高岭土或蒙托石、膨润土、硅藻土为载体,经捏合、挤条成型,350-400℃焙烧而成。但是上述脱氯剂同样都不适用于重整生成油的液相脱氯过程。
7.一般来说,碳质载体(如活性炭等)与活性金属组分的作用力较弱,其优点是可以提高活性金属组分的反应性能,但其不足是金属活性组分易于流失,特别是在液相反应条件下,流失更为严重,而液相物料中如果存在水,则流失会进一步加剧。此类载体用于脱氯剂时,表现为氯容减少,使用寿命缩短。
技术实现要素:
8.本发明的主要目的在于提供一种液相脱氯剂、其制备方法及应用,以解决现有技术中的液相脱氯剂氯容低的问题。
9.为了实现上述目的,根据本发明的一个方面,提供了一种液相脱氯剂,该液相脱氯剂包括:活性组分,活性组分选自a型分子筛、13x分子筛及nay分子筛中的至少一种,活性组分在液相脱氯剂中的重量含量为20%~40%;助剂组分,助剂组分选自氧化铜、氧化钠、氧化钾、氧化钡、氧化钙、氧化镁、氧化锌及氧化铁中的至少三种,助剂组分在液相脱氯剂中的重量含量为3%~10%;用于负载活性组分和助剂组分的载体,载体为碳粉,碳粉在液相脱氯剂中的重量含量为10%~33%;以及余量的粘结剂。
10.进一步地,粘结剂为硅溶胶和/或铝溶胶;优选粘结剂在液相脱氯剂中的重量含量为30%~55%。
11.进一步地,碳粉为平均孔径≤50nm的碳粉;优选碳粉的平均孔径为4~15nm,更优选碳粉的比表面积为200~300m2/g。
12.根据本技术的第二个方面,还提供了一种液相脱氯剂的制备方法,该制备方法包括:将助剂组分的前身物制成的溶液浸渍到碳粉上,得到第一混合物;将第一混合物干燥后与活性组分及粘结剂混合,得到第二混合物;将第二混合物进行挤条成型,得到脱氯剂前体;对脱氯剂前体进行焙烧,得到液相脱氯剂;其中,助剂组分选自氧化铜、氧化锌及氧化铁中的至少一种,活性组分选自a型分子筛、13x分子筛及nay分子筛中的至少一种。
13.进一步地,助剂组分的前身物为可溶性盐,优选为可溶性硝酸盐或可溶性醋酸盐。
14.进一步地,对脱氯剂前体进行焙烧,得到液相脱氯剂的步骤包括:对脱氯剂前体进行干燥,得到干燥前体;对干燥前体进行焙烧,得到液相脱氯剂。
15.进一步地,在80~120℃下对脱氯剂前体进行干燥,得到干燥前体,优选干燥的时间为3~5h。
16.进一步地,在氮气气氛下,对干燥前体进行焙烧,得到液相脱氯剂;优选地,焙烧的温度为350~650℃,焙烧的时间为1~6h。
17.进一步地,焙烧的温度为450~600℃,焙烧的时间为3~5h。
18.根据本技术的第三个方面,还提供了一种上述任一种液相脱氯剂在对重整生成油进行脱氯处理中的应用。
19.进一步地,应用是在重整生成油为液相的条件下进行脱氯处理,优选重整生成油无需脱水直接进行脱氯处理。
20.进一步地,脱氯处理的温度为30~80℃,质量空速为2~10h-1
,优选3~6h-1
;优选地,脱氯处理在0.1~3mpa的压力下进行;反应器高径比为2~8:1,优选为3~5:1。
21.应用本发明的技术方案,通过采用碳粉作为载体,碳粉吸附位较多,且孔容和比表面积大,有利于助剂组分和活性组分在活性碳粉上的负载,同时活性碳粉表面具有很多含氧官能团,提高了活性碳粉载体的极性,能够增加载体与活性组分间的结合强度,减少活性组分的流失以保持较高的氯容。同时氯化氢为极性分子,也更易于稳固地吸附在同样具有较大极性的高效活性碳粉载体的孔道结构中,进而有利于提高脱氯剂的脱氯精度。
具体实施方式
22.需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。下面将结合实施例来详细说明本发明。
23.术语解释:
24.氯容:氯容就是脱氯剂的承氯能力。
25.脱氯精度:即脱氯剂脱氯效果的一种体现,脱氯后油品氯含量越低说明脱氯剂的脱氯精度越高。
26.针对现有技术中的液相脱氯剂氯容低的问题,在本技术一种典型的实施方式中,提供了一种液相脱氯剂,该液相脱氯剂包括:活性组分、助剂组分、用于负载活性组分和助剂组分的载体以及粘结剂,其中载体为碳粉,碳粉在液相脱氯剂中的质量含量为10%~33%;活性组分选自a型分子筛、13x分子筛及nay分子筛中的至少一种,活性组分在液相脱氯剂中的质量比含量为20%~40%;助剂组分选自氧化铜、氧化钠、氧化钾、氧化钡、氧化钙、氧化镁、氧化锌及氧化铁的至少三种,助剂组分在液相脱氯剂中的质量含量为3%~10%;余量为粘结剂。
27.本技术所提供的液相脱氯剂,通过采用吸附位较多,孔容大(比如0.5~1.2cm3/g)和比表面积大(比如可以是150~600m2/g)的碳粉作为载体,有利于助剂组分和活性组分在活性碳粉上的负载,同时活性碳粉表面具有很多含氧官能团,提高了活性碳粉载体的极性,能够增加载体与活性组分间的结合强度,减少活性组分的流失以保持较高的氯容。同时氯化氢为极性分子,也更易于稳固地吸附在同样具有较大极性的高效活性碳粉载体的孔道结构中,进而有利于提高脱氯剂的脱氯精度(本技术的脱氯剂的脱氯精度能达到0.5ppm以下)。
28.本发明的液相脱氯剂对油品中无机氯以及有机氯中四氯乙烷、二氯甲烷的脱除具有良好效果,特别适用于重整生成油中氯化氢的脱除,同时适用于合成氨及甲醇等化工生产中的精脱氯,应用范围很广。
29.本技术所提供的液相脱氯剂中,活性组分选自上述类别的分子筛作为活性组分,具有有效脱除有机氯化物作用的有益效果。而在上述活性组分的基础上,选择氧化铜、氧化锌及氧化铁中的至少一种作为助剂组分,有助于提高活性成分的性能。粘结剂用于成型。上述各组分通过合理的用量配比,使得各组分的性能得以协同配合,进而使本技术的液相脱氯剂具有较高的氯容。
30.上述液相脱氯剂中,粘结剂用于对各组分进行粘结并使产品成型,其具体种类和用量可以在现有粘结剂的基础上合理调整得到。在一种优选的实施例中,粘结剂为硅溶胶或者铝溶胶;优选粘结剂在液相脱氯剂中的质量含量为30%~55%。选择硅溶胶或者铝溶胶作为粘结剂具有成本低,成型工艺简单的优势。优选硅溶胶为二氧化硅溶于硝酸溶液所形成的硅溶胶。优选铝溶胶为拟薄水铝石溶于硝酸溶液而形成的铝溶胶。其具体浓度可根据实际需要进行合理调整。
31.上述碳粉作为活性组分和助剂组分的吸附载体,为了更好地负载活性组分和助剂组分,最好具有较高的孔容和较大的比表面积。在一种优选的实施例中,碳粉为平均孔径≤50nm的碳粉;优选碳粉的平均孔径为4~15nm,更优选碳粉的比表面积为200~300m2/g。优选孔结构为微孔结构和介孔结构的碳粉,具有上述较大的孔容和比表面积,提高对活性组分和助剂组分的吸附能力,从而使其与载体的结合力较强而不易流失,从而保持较高的氯容。
32.在本技术第二种典型的实施方式中,提供了上述任一种液相脱氯剂的制备方法,该制备方法包括:将助剂组分的前身物制成的溶液浸渍到碳粉上,得到第一混合物;将第一混合物干燥后与活性组分及粘结剂混合,得到第二混合物;将第二混合物进行挤条成型,得到脱氯剂前体;对脱氯剂前体进行焙烧,得到液相脱氯剂;其中,助剂组分选自氧化铜、氧化锌及氧化铁中的至少一种,活性组分选自a型分子筛、13x分子筛及nay分子筛中的至少一种。
33.针对气液相分子扩散速率的差别和大分子烃特殊的分子结构,本技术的液相脱氯剂的上述制备方法,将碳粉浸泡在助剂组分前身物的可溶性盐中,经干燥处理后与活性组分a型分子筛、13x分子筛或nay分子筛混合,经粘结剂捏合成型后经过焙烧处理制得,其比表面积和孔容大,减少了分子扩散阻力对脱氯造成的影响。
34.而且,在焙烧处理过程中,助剂组分与载体接触的部分可以与碳元素发生部分还原反应,还原后与载体的碳粉发生更强的结合作用,类似于金属与碳的合金结构,其机理可能类似于金属材料领域的渗碳作用。这种结构与载体的作用力较强,因而加强了载体与活性组分之间的结合强度,减少了活性组分在使用过程中的流失。活性组分不流失是提高氯容量的最重要因素之一。
35.在一种优选的实施例中,助剂组分的前身物为可溶性盐,优选为可溶性硝酸盐或可溶性醋酸盐。氧化铜、氧化锌及氧化铁的前身物的可溶性盐分别为可溶性铜盐、可溶性锌盐及可溶性铁盐。根据实际需要进行合理选择。
36.上述制备方法中,焙烧的步骤可以在现有焙烧步骤基础上对焙烧条件进行适当调整得到。在一种优选的实施例中,对脱氯剂前体进行焙烧,得到液相脱氯剂的步骤包括:对脱氯剂前体进行干燥,得到干燥前体;对干燥前体进行焙烧,得到液相脱氯剂。先进行干燥再焙烧,有助于使焙烧的结构均匀。
37.上述焙烧前干燥的步骤中,干燥的方式不限。在一种优选的实施例中,在80~120℃下对脱氯剂前体进行干燥,得到干燥前体,优选干燥的时间为3~5h。
38.上述焙烧的步骤中,焙烧的具体温度可以合理调整以得到最适焙烧温度。在一种优选的实施例中,在氮气气氛下,对干燥前体进行焙烧,得到液相脱氯剂;优选地,焙烧的温度为350~650℃,焙烧的时间为1~6h。在一种更优选的实施例中,焙烧的温度为450~600
℃,焙烧的时间为3~5h。
39.在本技术第三种典型的实施方式中,提供了上述任一种液相脱氯剂在对重整生成油进行脱氯处理中的应用。本技术的液相脱氯剂对重整生成油的液相油品中无机氯和有机氯的脱除效果良好,可以将油品中的氯含量脱除至≤0.5mg/l。
40.在一种优选的实施例中,应用是在重整生成油为液相的条件下进行脱氯处理,优选重整生成油无需脱水直接进行脱氯处理。本技术的液相脱氯剂对重整生成油的液相油品中无机氯和有机氯的脱除效果良好,可以将油品中的氯含量脱除至≤0.5mg/l。因而无需脱水即可直接对重整生成油进行脱氯处理。
41.上述脱氯处理的具体参数条件可以根据重整工艺的具体过程及工艺要求确定。在一种优选的实施例中,脱氯处理的温度为30~80℃,质量空速为2-10h-1
,优选3-6h-1
;优选地,脱氯处理在0.1~3mpa的压力下进行;反应器高径比为2~8:1,优选为3~5:1。在其它条件一定的情况下,高径比越大,脱氯剂与油品接触时间越长,同时流体在床层分布更均匀,脱氯效果越明显,脱氯效率高。
42.下面将结合具体的实施例来进一步说明本技术的有益效果。
43.实施例1
44.将质量为6.0g拟薄水铝石(sb)粉加入到浓度为2mol/l、体积为60ml的硝酸溶液中,在室温下搅拌1h,作为粘结剂备用。
45.将质量为10.0g的碳粉(12%)加入到5.14g的mg(no3)2·
6h2o(以mgo计为0.80g)(助剂5.2%)、3.5g的ca(no3)2·
4h2o(以cao计为0.84g)以及5.0gcu(no3)2·
3h2o(以cuo计为1.70g)的水溶液中搅拌使其充分浸渍混合,其中的碳粉性质为:平均孔径为12.8nm,比表面积为200m2/g。
46.将45g粘结剂(54%)、碳粉混合物以及25g(30%)13x分子筛搅拌混合,然后将混合物捏合成型后,将样品置于干燥箱中在120℃中干燥4h,然后将其在550℃氮气氛围下的管式炉中稳定化处理4h,得到成品脱氯剂a。
47.实施例2
48.将质量为35.0g高分子二氧化硅颗粒加入到浓度为5mol/l、体积为60ml的硝酸溶液中,在室温下搅拌1h,作为粘结剂备用。
49.将质量为15.0g(13.4%)的碳粉加入到7.5g的nano3(以na2o计为2.74g)(助剂6.25%)、3g的ca(no3)2·
4h2o(以cao计为0.72g)、5.0g cu(no3)2·
3h2o(以cuo计为1.70g)及3.5g kno3(以k2o计为1.90g)混合水溶液中搅拌使其充分浸渍。其中的碳粉性质为:平均孔径为10.3nm,比表面积为220m2/g。
50.将多余的液体除掉后,将碳粉混合物与65g(58%)硝酸溶液与高分子二氧化硅颗粒的混合物以及25g(22%)13x分子筛搅拌混合。然后将混合物捏合成型后,将样品置于干燥箱中在80℃中干燥4h,然后将其在550℃氮气氛围下的管式炉中稳定化处理4h,得到成品脱氯剂b。
51.实施例3
52.将质量为20g高岭土加入到浓度为3mol/l、体积为60ml的硝酸水溶液中,在室温下搅拌45min,作为粘结剂备用。
53.将质量为10.0g(13.7%)的碳粉加入到6.5g的cu(no3)2·
3h2o(以cuo计为2.21g)
(助剂6.7%)、5.5g的ca(no3)2·
4h2o(以cao计为1.32g)、2.3g的ba(no3)2(以bao计为1.35g)的混合水溶液中搅拌使其充分浸渍。其中的碳粉性质为:平均孔径为8.5nm,比表面积为240m2/g。
54.将多余的液体除掉后,将碳粉混合物与30g硝酸溶液与高岭土的混合物及28g(38.4%)nay分子筛搅拌混合。然后将混合物捏合成型后,将样品置于干燥箱中在120℃中干燥4h,然后将其在550℃氮气氛围下的管式炉中稳定化处理3h,得到成品脱氯剂c。
55.实施例4
56.将质量为30g sb粉加入到浓度为3mol/l、体积为60ml的硝酸水溶液中,在室温下搅拌45min,作为粘结剂备用。
57.将质量为3.5g的mgso4·
7h2o(以mgo计为1.16g)(助剂7.4%)、5.5g的ca(no3)2·
4h2o(以cao计为1.32g)、7.0gcu(no3)2·
3h2o(以cuo计为2.38g)、5.2g的nano3(以na2o计为1.90g)配成混合水溶液,加入质量为30.0g的碳粉(33%)、34g上述粘结剂(37.5%)以及20g(22%)a型分子筛,搅拌使其中的盐充分浸渍到碳粉上。其中的碳粉性质为:平均孔径为7.8nm,比表面积为255m2/g。
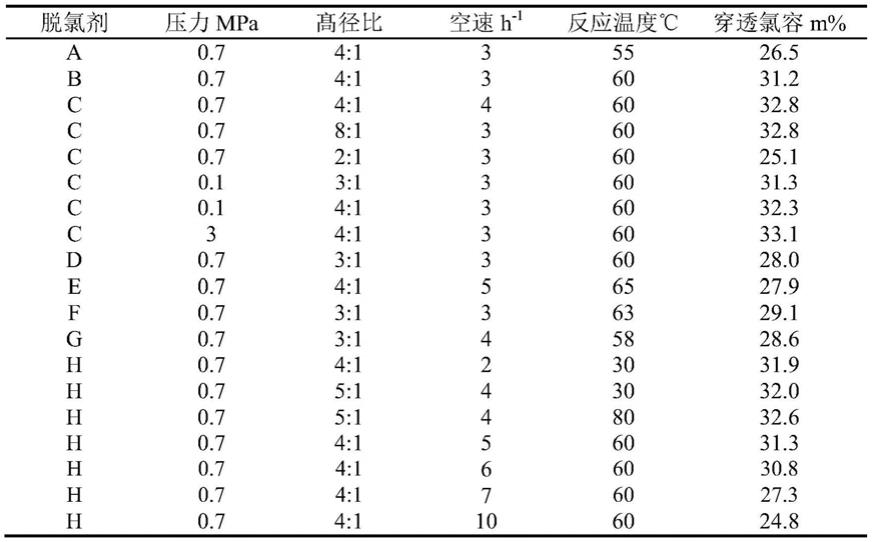
58.经过滤后将样品置于干燥箱中在120℃中干燥4h,然后将其在550℃氮气氛围下的管式炉中稳定化处理4h,得到成品脱氯剂d。
59.实施例5
60.将质量为30g sb粉加入到浓度为3mol/l、体积为60ml的硝酸水溶液中,在室温下搅拌45min,作为粘结剂备用。
61.将质量为3.6g的fe(no3)3·
9h2o(以fe2o3计为0.70g)(助剂5.2%)、3.5g的ca(no3)2·
4h2o(以cao计为0.84g)、2.8g的kno3(以k2o计为1.30g)、5.0g的cu(no3)2·
3h2o(以cuo计为1.70g)配成水溶液,加入质量为15.6g的碳粉(17.7%)、48g上述粘结剂(54.5%)以及20g 13x型分子筛(22.7%),搅拌使其中的盐充分浸渍到碳粉上。其中的碳粉性质为:平均孔径为6.6nm,比表面积为268m2/g。
62.经过滤后将样品置于干燥箱中在90℃中干燥5h,然后将其在600℃氮气氛围下的管式炉中稳定化处理2h,得到成品脱氯剂e。
63.实施例6
64.将质量为30g sb粉加入到浓度为3mol/l、体积为60ml的硝酸水溶液中,在室温下搅拌45min,作为粘结剂备用。
65.将质量为3.6g的fe(no3)3·
9h2o(以fe2o3计为0.70g)(5.2%)、4.5g的nano3(以na2o计为1.64g)、6.0gcu(no3)2·
3h2o(以cuo计为2.04g)、3.5g的mg(no3)2·
6h2o(以mgo计为0.55g)配成混合水溶液,加入质量为22.0g的碳粉(23.4%)、43g(45.8%)上述粘结剂以及24g nay分子筛(25.6%),搅拌使其中的盐充分浸渍到碳粉上。其中的碳粉性质为:平均孔径为6nm,比表面积为270m2/g。
66.经过滤后将样品置于干燥箱中在85℃中干燥5h,然后将其在450℃氮气氛围下的管式炉中稳定化处理5h,得到成品脱氯剂f。
67.实施例7
68.将质量为30g sb粉加入到浓度为3mol/l、体积为60ml的硝酸水溶液中,在室温下搅拌45min,作为粘结剂备用。
69.将质量为3.6g的fe(no3)3·
9h2o(以fe2o3计为0.70g)(10.7%)、4.5g的nano3(以na2o计为1.64g)、4.5g的kno3(以k2o计为2.09g)、6.0g的cu(no3)2·
3h2o(以cuo计为2.04g)配成混合水溶液,加入质量为6.0g(10%)的碳粉、30g上述粘结剂(50%)以及18g(30%)a型分子筛搅拌使其中的盐充分浸渍到碳粉上。其中的碳粉性质为:平均孔径为5.5nm,比表面积为280m2/g。
70.经过滤后将样品置于干燥箱中在95℃中干燥4h,然后将其在650℃氮气氛围下的管式炉中焙烧1h,得到成品脱氯剂g。
71.实施例8
72.将质量为30g sb粉和高分子二氧化硅颗粒加入到浓度为1mol/l、体积为90ml的硝酸溶液中,在室温下搅拌50min。
73.将质量为15.0g的碳粉加入到由质量为7.5g的mg(no3)2·
9h2o(以mgo计为1.18g)、2.3g的ba(no3)2(以bao计为1.35g)、6.5gcu(no3)2·
3h2o(以cuo计为2.21g)配成的30ml水溶液中,搅拌使其充分浸渍。其中的碳粉性质为:平均孔径为4.3nm,比表面积为300m2/g。
74.将多余的液体除掉后,将碳粉混合物、15gx型分子筛加入到30g硝酸溶液与sb粉和高分子二氧化硅颗粒混合物中搅拌混合。预处理好的混合物在捏合机中捏合成型,将样品置于干燥箱中在120℃中干燥3h,然后将其在350℃氮气氛围下的管式炉中稳定化处理6h,得到成品脱氯剂h。
75.实施例9
76.本实施例为脱氯剂a-h脱除重整生成油中hcl性能的测试结果。将氯含量为5μg/l、水含量为20μg/l的重整生成油在30~80℃,0.1~3mpa、空速为2~10h-1
的条件下装填脱氯剂进行液相动态脱氯实验,测定流出脱氯罐的重整生成油中的氯含量,当氯含量大于0.5μg/l时,即可视为穿透,分析脱氯剂的穿透氯容。将穿透后的样品从脱氯装置中卸下后进行氯容测定,其结果如表1。
77.氯容的测定方法及计算公式如下:
78.穿透氯容=穿透后脱氯剂中氯的质量含量/穿透后脱氯剂总质量*100%。
79.表1:样品的氯容
[0080][0081]
从上表1可以看出:脱氯剂a-h都具有良好的氯容。在相同的条件小,压力从0.1-3mpa、温度30-80℃范围内,脱氯剂的氯容变化不大,表明压力与温度对脱氯的影响很小;空速从2-10范围内,当空速大于7之后,氯容由6时的30.8%很快下降到27.3%,氯容明显下降,说明较好的空速范围为3-6;髙径比在2:1~8:1的范围内,氯容随高径比的增加而增加,当髙径比为2时,氯容为25.1%,当髙径比为3时,氯容为31.3%,增加的很高,当髙径比增加到8时,氯容为32.8%,增加不再明显。髙径比不是越高越好,髙径比过高,床层的压力降越大,因此,更优选的髙径比为3:1~5:1之间。
[0082]
比较例1
[0083]
参考cn1088388c实施例1的方法,按重量百分数caco335%、znco
3 20%及高岭土20%,将三种物质混在一起,然后在混合样中加入25%na2co3,混匀后挤条成φ0.45-0.9mm条,干燥、焙烧1小时后得到脱氯剂样品。
[0084]
比较例2
[0085]
参考cn103127903a实施例2的方法,称取25g的硫酸亚铁,40g的氧化钙,15g活性凹凸棒石粘土混合,之后进行混捏45min;将上述均匀后的干料加入硅溶胶35g,将上述物质湿混30min,直到混碾成膏状物,将上述膏状物放在小型挤条机上,挤出φ4条型物;将上述条状物在180℃进行干燥2小时制得脱氯剂样品。
[0086]
比较例3
[0087]
按实施例8的方法和过程,其中的稳定化处理改变为常规的焙烧处理,即在400℃焙烧3小时,得到最终脱氯剂。
[0088]
将样品h与比较例1、2和3中得到的样品在相同条件下,分别对水含量为20μg/l、氯含量为5μg/l的重整生成油和氯含量为5μg/l、水含量为0.5μg/l的重整生成油进行脱氯试验。温度选为30℃,髙径比为4:1,液空速为4h-1
,压力为3mpa,得出的氯容数据见表2。
[0089]
表2实施例8与比较例1-3的脱氯剂氯容随水含量变化
[0090][0091]
从表2可以看出,油品中水含量越高,脱氯剂氯容就越小;相比其它的脱氯剂,微量水含量从0.5增加到25μg/l对实施例8中的脱氯剂的氯容从35.7减少到31.8%,减少了约4个百分点,而对比例脱氯剂的氯容下降了10多个百分点,表明本发明优化后的脱氯剂具有很好的抗水性。
[0092]
从以上的描述中,可以看出,本发明上述的实施例实现了如下技术效果:本发明液相脱氯剂原料成本低制备方法简单,而且脱氯剂的使用寿命长,机械强度大,各组分在液相脱氯条件下稳定,不易流失,同时该脱氯剂同时具有一定的脱硫、脱氮效果,有利于脱氯物料的后续处理过程。
[0093]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。